基于MST的基因数据社团挖掘算法
2014-01-15刘飞
刘飞
(宝鸡文理学院 物理与信息技术系,陕西 宝鸡 721016)
随着高通量测序技术的发展,产生了大量的基因表数据。这就需要一些方法来分析这些数据,得出这些数据包含的潜在信息[1]。因此对大量的基因表示数据和微生物网络进行社团挖掘,有效地鉴别基因表示数据的模式是研究DNA序列的重要基础。利用统计学、生物信息学和计算机科学提供一些理论和方法,解决生物信息学中海量微生物数据的计算和信息处理问题越来越受到人们的重视。人们发现许多真实网络中都存在着一个重要的特性——社团(即模块)结构,即整个网络是由若干个社团构成的,这些社团具有内紧外松的结构特征[2-4]。例如在生物网络中可根据各个基因节点不同的功能特性划分为不同的社团,每个社团内部节点间连接相对紧密,各个社团之间的连接却相对稀疏。
现在有很多用于处理基因表达数据社团挖掘的算法,如Eisen等人[5]应用分层的平均边社团挖掘算法对酵母基因网络进行分析;Ben-Dor和Yakhini等人[6]开发的CAST算法;K-平均值算法;自组织映算法;主成分分析算法等。使用不同的数据分析技巧和不同社团挖掘算法对同样一个基因数据集会产生不同的效果。这些算法被实践证明性能确实非常优越,但也存在一些不足,很多算法没有证明自己的社团分割是否是最优的。本文我们介绍一种基于最小生成树(minimum spanning trees,MST)的算法,用于处理基因表数据的社团挖掘。这种算法将基因表达数据的模块挖掘问题转换为树的分割问题,通过删除树中一些特定意义的边,将最小生成树分成若干个子树,每个子树就是一个社团模块。本文通过实验证明了该社团模块挖掘算法的优越性。
1 最小生成树的相关定义
设G=(V,E,W)是一个连通图,V是G中点的所有集合,E是G中边的所有集合,W是边的权值。一个图的生成树指的是它的最小连通图,包含所有顶点,图中边的个数比点的个数少一,如若再多一条则会形成回路。而最小生成树[7]指的是所有生成树中各边权值之和最小的那棵生成树,根据最小生成树的性质,普里姆(Prim)算法和克鲁斯卡尔(Kruskal)算法可以非常便捷地计算出一个连通图的最小生成树。下面分别介绍这两种算法:
普里姆算法,假设N=(V,{E})是连通网,TE为最小生成树中边的集合。
1)初始化一个新的图,图中包含原连通图的所有节点,但是没有边,即U={u0}(u0∈V),TE=Φ;
2)在所有u∈U,v∈V-U的边中选一条权值最小的边(u0,v0)并入集合 TE,同时将 v0并入 U;
3)重复上述步骤2),当U=V时循环结束。
此时,TE 中必含有 n-1条边,则 T=(V,{TE})为 N 的最小生成树。
克鲁斯卡尔算法,假设N=(V,{E})是连通网,将网中所有的边按照权值从小到大排序:
1)将连通网中的每一个顶点看成一个集合,则有n个集合;
2)从权值最小的边开始选取,所选边的顶点处在不同的两个集合中,把这条边放到生成树边的集合中,并把这条边的两个顶点合并。
3)重复上述步骤2),当所有的顶点并入一个集合时,结束操作。
可以看出,普利姆算法逐步增加U中的顶点,可称为“加点法”。克鲁斯卡尔算法逐步增加生成树的边,与普里姆算法相比,可称为“加边法”。利用Prim算法或者Kruskal算法可计算出一个连通网络的最小生成树如图1所示。
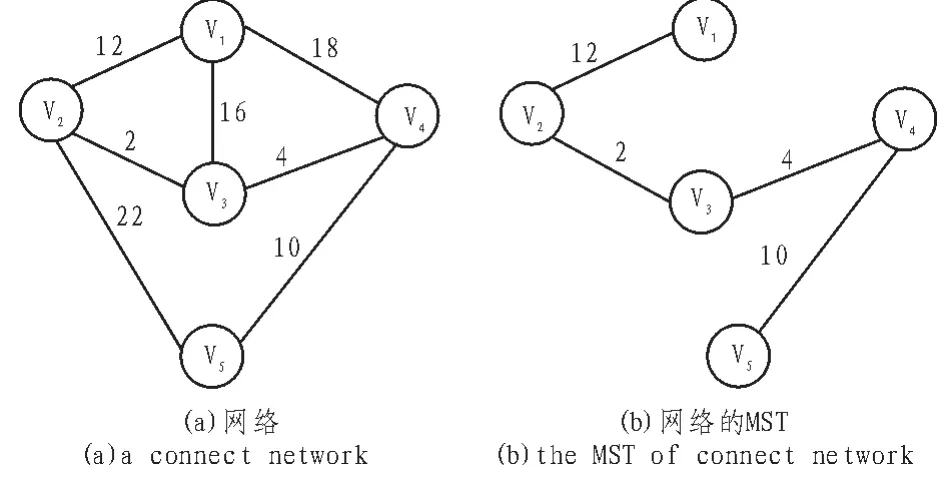
图1 一个连通网络及其MSTFig.1 A connect network and its minimum spanning tree
2 基因表达数据的生成树表示方法
使用最小成生树来表示一个基因表达数据集,把多维基因表示数据的社团模块挖掘问题转化为最下生成树的分割问题。 权图G(D)=(V,E),其中点集合V={didi∈D },di=(di1,di2,…,dim)表示i个基因,每个基因由m个属性。边集E={di,dj对于 di,dj∈D 且 i≠j}(这里的边集可以表示基因相似或相异程度的一个度量)。显然权图G(D)=(V,E)是一个完全图,图中每一条边(u,v)∈E可以表示为两个基因之间的相似程度或相异程度 ρ(u,v),u和 v之间的权值 ρ(u,v)可采用皮尔逊相关系数或者欧几里德距离等其他距离度量方法。下面分别介绍这几种方法,其中d¯i表示di的平均值。
1)皮尔逊相关系数
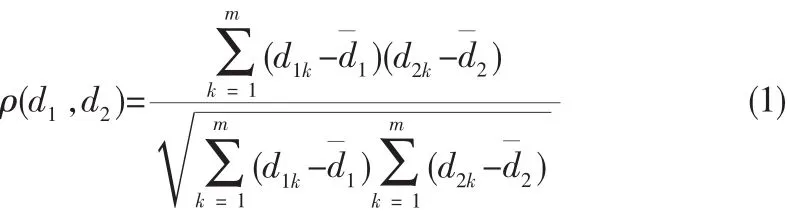
2)欧几里德距离
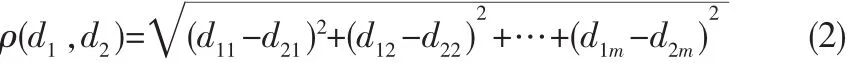
3)斯皮尔曼相关系数
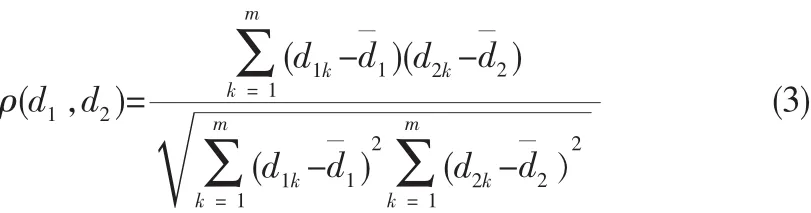
图2(a)是一个基因数据的完全网络,它们之间的边可以通过上述方法计算得到,图2(b)是其用Prim算法或者Kruskal算法计算得出的最小生成树,从图中可以看出分成了3个社团,同一社团中的数据点用较短的树边连接,而不同社团间的数据点由长的树边连接。在一个社团内部相邻点之间的距离小于不同社团之间点的距离。那么通过清除G(D)的最小生成树中具有最大距离的S-1条边,网络就可以分成S个社团。
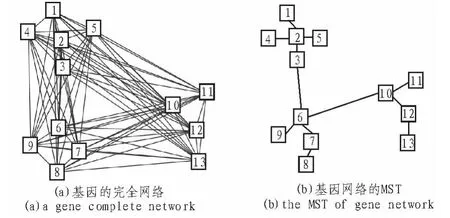
图2 一个基因的完全网络及其最小生成树Fig.2 A gene complete network and its minimum spanning tree
3 基于MST的社团挖掘算法
不同的社团挖掘算法需要不同的评价准则函数,为了获得最佳的社团划分结果,在这里将叙述了3种评价准则函数和它们对应的社团挖掘算法。
1)去除MST长边的社团挖掘算法
一个最为简单的评价准则函数就是去掉MST中S-1条长边,而形成S个子树,使得所有子树的边权值之和最小。这个评价准则函数的根据如下:如果两个点之间的权值很小,则它们应该属于同一社团(子树),反之亦然。但是当不同社团之间的连接边的权值很小时,或者存在一些噪声或者孤立点时,这种判断方法就不尽理想了[8]。为了避免这种情况,可在此社团挖掘算法划分中判断新的社团是否为孤立点,通过消除孤立点来提高这种社团挖掘算法的精度。
2)重复社团挖掘算法
把所得的最小生成树(MST)分割成S个子树Ti{,它所依据的评价准则函数如下:
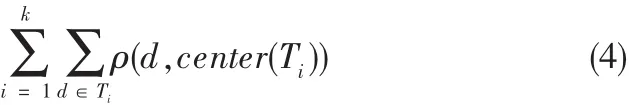
重复社团挖掘算法使得任意一个社团的中心和团内基因点之间边的权值之和最小,用不同的度量方法时,中心center(Ti)会有不同的值。此外我们还可以采用平方误差评价准则函数,其评价准则函数如下:
重复社团挖掘算法以任意一个最小生成树的S分割开始,首先计算每一个子树的中心值,然后重复下面步骤:把相邻的两个社团(一般通过长边相连)合并为一个社团,在新的社团中去除所有的边,通过评价准则函数(4)来进行最优二社团划分。
3)改进的重复社团挖掘算法
改进的重复社团挖掘算法是一种面向中心的社团划分方法,同样以任意一个最小生成树的S分割开始,并计算每一个子树的中心值 center(Ti),i=1,2,…,S。 删除最小生成树中的所有边,对于最小生成树中的任意结点dj,计算
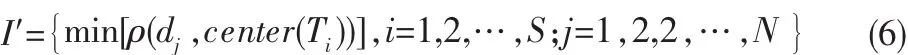
当 I′值最小时的 I=i,即 dj距离 center(Ti)最近,那么 dj应该属于子树Ti中的结点。与评价准则函数(4)相比,这个改进的重复社团挖掘算法更容易获得全局最优解。
4 实验仿真
实验用的仿真数据为Sherlock等人[9]的基因表达数据,这个数据集包含500多个基因,每个基因有18个属性。使用第2个社团挖掘算法,欧几里德距离作为距离度量。图3显示采用第2种算法时最佳S社团挖掘值对社团数目S的关系,可以看到当S从1到6时社团挖掘值显著改善,之后随着S值增加改善率下降。因此整个基因数据集分为6个社团比较理想。

图3 基因表达数据的准则函数与社团数目对照图Fig.3 The chart of criterion function and community number for gene expression data
随机生成80个数据,每个数据有20个属性,构造它的最小生成树并使用上面的社团挖掘算法进行社团划分。用重复社团挖掘算法和改进的重复社团挖掘算法对S选择不同值时,计算出它们总的误差平方和如图4所示。可以看出改进后的社团挖掘算法性能有一定提升,当S为5时得到最佳的分团结果。
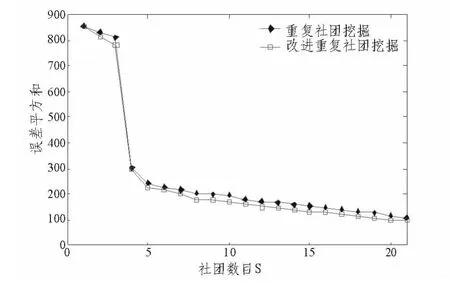
图4 不同方法基因表达数据的准则函数与社团数目对照图Fig.4 The chart of criterion function and community number with different methods for gene expression data
5 结束语
针对一些生物网络的社团划分问题,人们提出了很多行之有效的方法。然而使用MST表示多维基因表达数据集以解决基因表示数据的社团划分问题,确实是一种严格且有效的方法,特别对于一些准则函数,这一算法能保证获得全局最优解。将多维数据社团划分问题转换为最小生成树的分割问题,可以解决各种生物学分析问题,如动植物系统分类、生物序列的特征识别、蛋白质家族分类等。同一社团内由类似的基因数据组成,研究和分析每个社团的结构和功能以及社团之间的关系,这对深刻认识诸多生物过程的本质有重要意义。
[1]Eric S lander.Array of hope[J].Nature Genetics,1999(21):3-4.
[2]Girvan M,Newman M E J.Community structure in social and biological networks[J].Proceedings of the National Academy of Sciences,2002,99(12):7821-7826.
[3]王艳,李应兴,靳二辉.复杂网络健壮社团挖掘算法[J].计算机工程与应用,2012,48(31):36-39.WANG Yan,LI Ying-xing,JIN Er-hui.Novel algorithm for robustcommunity in complex networks [J].Computer Engineering and Applications,2012,48(31):36-39.
[4]Brandes U,Delling D,Gaertler M,et al.On modularity clustering [J].Knowledge and Data Engineering,IEEE Transactions on,2008,20(2):172-188.
[5]Eisen M B,Spellman P T,Brown P O,et al.Cluster analysis and display ofgenome-wide expression patterns[J].Proceedings of the National Academy of Sciences,1998,95(25):14863-14868.
[6]Ben-Dor A,Shamir R,Yakhini Z.Clustering gene expression patterns[J].Journalofcomputationalbiology,1999,6(3-4):281-297.
[7]Frank Dehne,JohnIacono,Jorg-Rudiger Sack.Algorithms and Data Structures [M].Springer-Verlag Berlin and Heidelberg GmbH&Co.K,Edition.2011.
[8]Ramonell K M,Zhang B,Ewing R M,et al.Microarray analysis of chitin elicitation in Arabidopsis thaliana[J].Molecular Plant Pathology,2002,3(5):301-311.
[9]Sherlock G.Analysis of large-scale gene expression data[J].Current Opinion in Immunology,2000,12(2):201-205.
