大孔Mo-Ni/Al2O3催化剂的催化裂化柴油加氢性能研究
2014-01-02王鼎聪赵德智
丁 巍 王鼎聪 赵德智 柯 明
(1中国石油大学(北京)重质油国家重点实验室,北京 102249)
(2中国石化抚顺石油化工研究院,抚顺 113001)
(3辽宁石油化工大学化学化工与环境学部,抚顺 113001)
在人们对于生存环境日益重视的今天,各国环保法规对车用燃料中导致污染排放的硫、氮、芳烃等杂质含量的限制日趋严格[1-2]。世界原油重质化的趋势逐渐加大,原油价格逐步上升,催化裂化技术处理重质油的市场份额逐渐增加。为生产超低硫、低芳烃的清洁柴油,研究者们采取了很多措施来脱除油品中的硫化物[3-6]、氮化物[7]和多环芳烃[8]。其中,催化裂化柴油含有高含量、难脱除的芳烃,这些多环化合物是胶质的前驱体,在加氢处理过程中极易结焦,使催化剂无法应用在这种劣质油品加氢处理中。一般加工过程都采用催化裂化柴油与直流柴油进行复配处理,制备单独加工催柴的催化剂显得尤为重要。加氢技术是改善油品质量非常有效的手段,而采用高活性的加氢催化剂是最有效、投资最省的途径之一。提高加氢催化剂活性的一个重要手段是增加活性中心的数量。
加氢精制催化剂的性能受载体、活性组分及助剂的影响。三者各有各的主要作用,但它们不是孤立分割的,而是相互渗透相辅相成的[9]。载体在催化剂中起担载活性组分、提高活性组分和助剂分散度的作用,在一定程度上也参与了某些反应[10]。随着研究的深入,现有开发的加氢催化剂载体有γ-Al2O3[11]、TiO2[12]、碳[13]、ZrO[14]、介孔分子筛[15]或沸石[16]等。 其中,γ-Al2O3具有价格较低,再生性能好等特点,广泛应用于汽柴油加氢处理上。自组装合成γ-Al2O3的方法在近几年来广泛的被应用于加氢催化剂的制备中[17-18],独特的制备技术可以增加负载量,大大地改善加氢反应活性。一般工业上重油加氢处理过程广泛应用的氧化铝载体催化剂的孔径在3~10 nm,孔容在0.6~0.9 mL·g-1左右。对于劣质催柴中的多环芳烃、胶质、沥青质及沥青质胶团来说,在10 nm左右的孔道中的扩散,会受到一定程度的扩散阻力,从而影响加氢效果。本实验组利用自组装方法合成的纳米自组装催化剂成功的应用在了劣质柴油的加氢处理上[19-20],处理效果良好。
本文将以二次纳米自组装大孔氧化铝载体为主体,以钼镍活性金属为客体,采用三次纳米自组装合成法制备高活性金属中心的主客体催化剂。分析了主体及催化剂的孔结构,并评价了其加氢性能。
1 实验部分
1.1 药品及仪器
1.1.1 实验药品
九水硝酸铝,分析纯;尿素,分析纯;三乙醇胺,分析纯;聚异丁烯马来酸三乙醇胺酯,自制;150HVI(润滑油基础油),工业品;去离子水,自制;顺丁烯二酸酐,磷酸,三氧化钼,碱式碳酸镍,分析纯;催化裂化柴油。
1.1.2 实验仪器
美国MICROMERITICS公司生产的ASAP 2420物理吸附仪,用于催化剂的比表面积、孔容、孔径及孔分布;在日本理学生产D/2500型X-射线衍射仪上测出样品XRD晶相结构,光源Cu Kα射线波长0.154 nm,管工作电压和电流分别为40 kV和80 mA;日本生产JSM-7500F型扫描电镜,加速电压20 kV,用于观察纳米自组装体的外貌及粒子的大小;JEOL公司生产JEM2100型透射电镜,点分辨率0.23 nm,用于观察纳米自组装体颗粒形貌、分散状况和粒径大小及其分布状况;德国NETZSCH公司生产的DSC204HP高压差热分析仪,用于一定压力下物质与参比物之间能量差随温度的变化及固体和液体样品在恒温和升温过程中的物相转变和热焓变化测量。
1.2 主客体催化剂的制备
将0.001~0.01 mol·L-1聚异丁烯马来酸三乙醇胺酯与0.07~0.2 mol·L-1的润滑油基础油在100℃下搅拌混合,然后加入 1~5 mol·L-1尿素和 1 mol·L-1九水硝酸铝制得一级超增溶胶团自组装体,在100℃反应3 h,产物经水洗、干燥,得到二级纳米自组装体,压条成型,150℃烘干,600℃焙烧6 h,得到大孔容氧化铝载体。将制备好的纳米自组装载体用等体积法均匀浸渍钼镍磷溶液,然后110℃烘干,450℃焙烧,得到本实验纳米自组装主客体加氢处理催化剂。
1.3 催化剂活性评价
加氢反应在微型反应器中进行,催化剂用量为15 mL(按其体积计)。进行加氢反应前用质量分数为2%的硫化油进行预硫化,在230℃下将催化剂(三次纳米自组装主客体催化剂或参比剂)预硫化4 h,再从230℃加热到360℃,在360℃停留4 h,液时空速为2.0 h-1,预硫化共15 h。然后在此温度下,压力 7.0 MPa,空速 1.5 h-1,氢油比(体积比)为 600∶1 的反应条件下,对镇海炼化催化裂化柴油的脱硫、脱氮及芳烃饱和能力进行评价。其原料油的性质见表1。

表1 催化裂化柴油的性质Table 1 Properties of the catalytic cracking diesel fraction
2 结果与讨论
2.1 纳米自组装载体的表征
2.1.1 SEM分析
图1中(a)是纳米氧化铝自组装载体的SEM图,图中清楚可见大量直径在100~200 nm的棒状纳米氧化铝粒子。他们以松散的交叉堆积方式搭建出具有不同直径大小的堆积孔道。从几何学的角度来看,采用常规方法制备的氧化铝载体直径在10~20 nm范围内,他们堆积的间隙孔道的最可几孔径在20 nm以下,而采用纳米自组装方法制备的大于100 nm直径的棒状体堆积的孔道应大于30 nm[21]。除了交叉堆积外,从图1(b)中还可以看到一些纳米棒状体呈平行状态生长,这一结果,主要是由于95%熔盐增溶物占据了大量的体积与氧化铝形成自组装体,模板剂控制自组装反应的进行,使γ-Al2O3在最少的面积上形成紧密堆积的结构,这种框架结构具有贯穿性,有利于动力学扩散[22,23]。

图1 载体的SEM图像Fig.1 SEM images of the support
2.1.2 热重分析

图2 载体的TG/DSC曲线Fig.2 Curve of TG/DSC of the support
图2中失重曲线(TG)说明二次纳米自组装氢氧化铝分解经历3个过程,分别为268、390、625℃。在268℃时,失重56.78%,这是因为拟薄水铝石失水转化成氧化铝和部分有机物的挥发及氢氧化物的分解造成的;390℃,失重65.62%,部分有机模板剂脱除;当焙烧到625℃时,纳米自组装氢氧化铝失重高达73.79%,再升高温度到808℃,失重不明显,说明模板剂及水等物质几乎已完全被脱除。结合DSC曲线可以看出,612℃以后已完全形成γ-Al2O3晶相,在此温度下焙烧,大部分模板剂被脱除,会使聚合的氢氧化铝体积发生急剧收缩,产生大孔。其孔容可达到1.77 cm3·g-1,孔隙率高达93%[24]。这是造成SEM中大孔道形成的原因。
2.2 载体与催化剂孔结构性质分析
催化剂FA-40是基于二次纳米自组装载体为主体,经三次纳米自组装法制备的主客体催化剂。参比剂F-5是以普通氧化铝为载体,浸渍法制备的工业用柴油加氢精制催化剂。
表2中FA-40的压汞数据表明,其孔容、孔径较大,在6 nm和40 nm左右出现双峰孔结构,与氮吸附法测定的结果相似(见图3a),这种孔结构为催化反应提供了充足的活性中心,同时也为反应物和产物提供了扩散的孔道。高达35.70%的活性金属量,使主体的比表面积减小到114 m2·g-1,堆积密度增加0.22 cm3·g-1,但是孔径尺寸变化不大。而F-5的孔容、孔径及金属含量都比FA-40小,虽然比表面积较大,但是其堆积密度大约为FA-40的2倍。说明F-5存在大量的小孔结构,且20.14%的活性金属以单层分散的形式负载在载体上,形成7.0 nm的单峰孔结构。这是由于普通载体的孔结构限定了金属的负载量以及制备方式决定了金属在孔道中的分散形式。另外,表2中计算结果表明,FA-40和F-5的单位体积活性金属含量几乎相同,为200 g·L-1。如果保证相同的加氢活性,显然FA-40具有更高的质量空速,从而增加处理量,间接的降低了催化剂的成本。
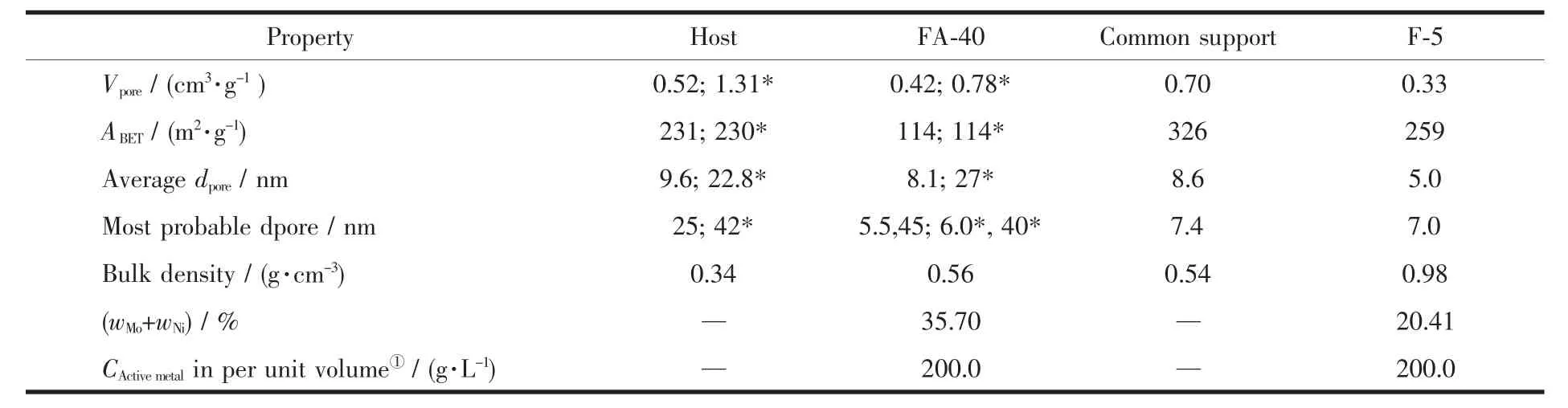
表2 载体与催化剂孔结构性质比较Table 2 Comparison of the pore structure characteristic for the supports and catalysts

图3 载体与催化剂的孔分布Fig.3 Pore size distribution of the different supports and catalysts
图3(b)也可以看出,纳米自组装载体和催化剂的氮气吸附法与压汞法测得的孔大部分集中在10~100 nm之间。压汞法数据表明,FA-40在此区间的孔分布约占80%的比例,>100 nm的孔分布也有12%。氮吸附数据表明,纳米自组装载体和FA-40在小于10 nm的孔分布比例为37.02%和59.89%,证明了大量小孔的存在。此范围内催化剂比载体的孔分布比例上升了22.87%,说明高达35.7%的活性金属在共浸剂的作用下已成功地自组装成不同形状的纳米粒子且松散地相互堆积成大量的间隙孔。
对于F-5来说,载体在10~30 nm的孔分布占总体的57.88%,由于金属的负载,使此范围的孔分布比例急剧下降到15.48%,同时,在小于10 nm的范围内高度集中,<6 nm和6~10 nm的孔分布分别占42.68%和38.8%,占总孔分布的81.48%;>30nm的孔分布没有太大变化。
以上证明了不同的制备工艺对载体和催化剂的孔性质、孔结构以及金属的负载量都有很大影响。
2.3 XRD分析
图4可以看出载体和催化剂的晶相结构,(a、b)为载体,在 2θ=37.6°,46.1°和 67.0°处,明显出现了γ-Al2O3的(311)、(400)和(440)3 个晶面的特征衍射峰,两种载体的晶相很相似。但自组装载体在37.6°处峰较宽,说明有大的纳米粒子形成。(c)在2θ=18.15°、24.2°和 27.38°处是 MoO3的特征吸收峰,27.38°处峰型较宽,再次证明Mo物种的活性组分是以纳米粒子形式存在,(d)在2θ=18.5°处也有一小峰,但很不明显,说明Mo物种向高能级扩散或形成无定形或非晶态粒子存在于催化剂中。

图4 载体和催化剂的XRD谱图Fig.4 XRD patterns of the supporter and catalysts
2.4 TEM分析
图5给出催化剂和参比剂的微观透射扫描电镜图像。图a可以看到直径小于2 nm金属粒子均匀分布在大孔Al2O3主体上,尽管分布密集,但颗粒互相之间并没有聚集。说明这些金属粒子均以形成纳米粒子。这是在自组装合成过程中活性金属以螯合物的形式高度分散在大孔Al2O3主体表面,经焙烧脱去共浸剂后,这种良好的结构被保持,成为均匀分散的纳米粒子。而F-5载体的粒径较小,一般在10~30 nm之间,在浸渍过程中,活性金属均以单层或多层分散的形式吸附在孔道内,过量的溶质沉积在小孔内,导致小孔堵塞,介孔变小。所以图b中会出现许多细小的金属黑色条型和黑点。从图c和d的金属硫化态可以看出,FA-40的金属硫化层数3~4层,而F-5的层数大多为1~2层,所以前者的活性位更多。
2.5 催化剂的活性评价
以催化裂化柴油为原料,在微型反应装置上反应20 h对催化剂的加氢脱硫、脱氮及芳烃饱和加氢性能进行比较,结果见图6。对FA-40进行300 h的加氢实验,结果见图7。
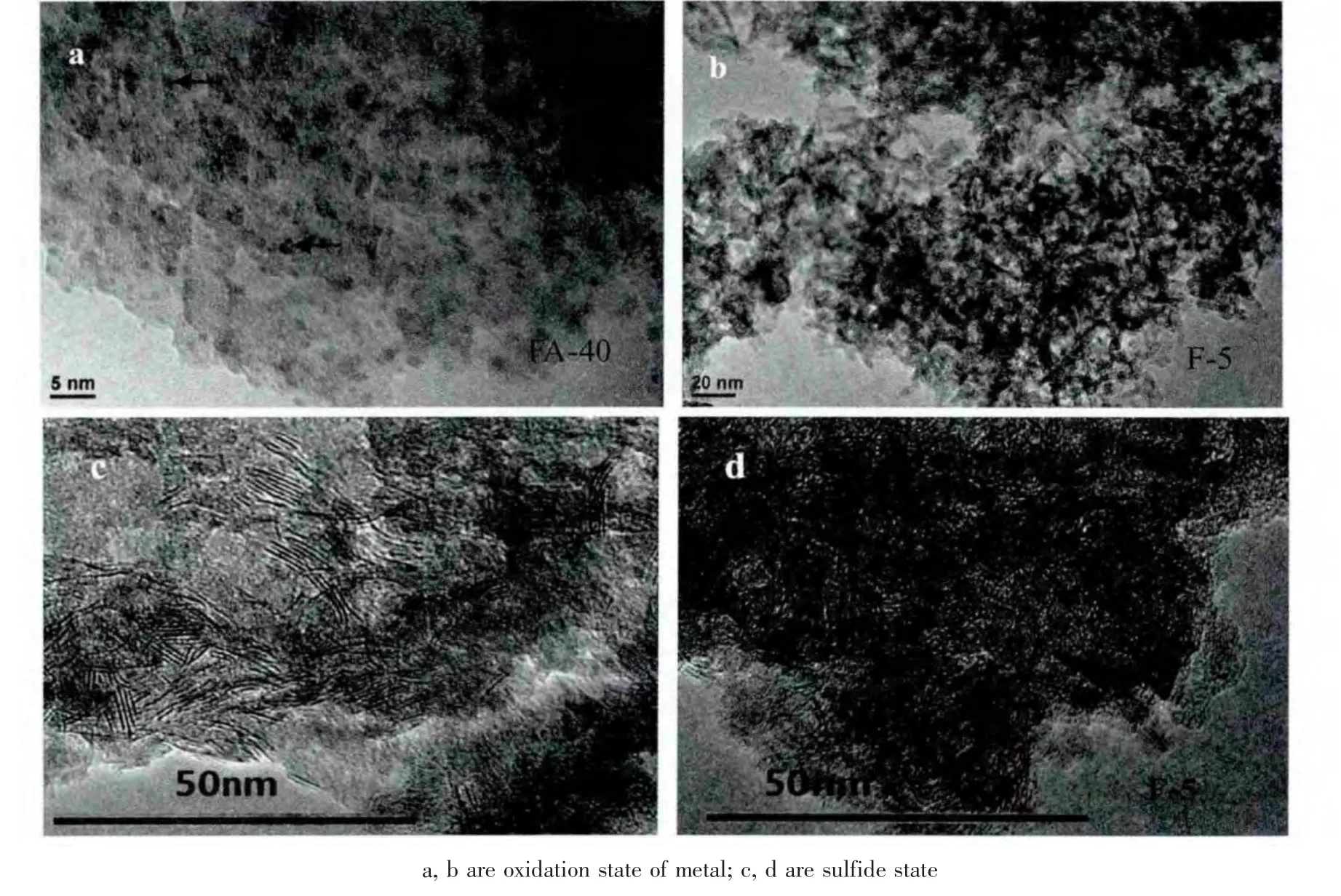
图5 不同催化剂的TEM照片Fig.5 TEM images of the difference catalysts

图6 加氢性能随时间变化曲线Fig.6 Curve of changes for hydrogenation performance with time
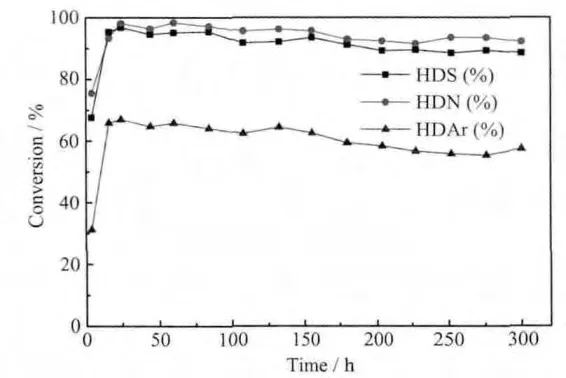
图7 催化剂FA-40随时间变化的加氢性能Fig.7 HDS,HDN and HDAr conversion with on stream over FA-40 catalyst
从6图中看出,在刚刚开始反应4 h时,两个催化剂的脱硫、脱氮率都较高,对于FA-40来说,杂质的转化率随时间的变化逐渐增大,到10 h后趋于稳定;而F-5恰恰相反,8 h以后转化率下降迅速,到12 h时脱硫率和芳烃饱和率降为80.9%、31.3%,之后趋于稳定;脱氮率仍在继续下降,当16 h时降到54.6%。原因是反应开始时,参比剂的小孔结构较多活性金属分布均匀,提供了较快的反应速率和催化活性,随着反应的进行,脱氢和缩合反应严重,导致积碳的生成,生成的焦炭使大量小孔堵塞,所以转化率急剧下降。而FA-40因为大孔的存在,为反应物和产物提供了扩散的孔道而抑制了结焦的产生,使加氢反应顺利进行。在20 h时,FA-40的脱硫、脱氮及芳烃饱和率分别达到94.4%、95.5%和67.9%,与F-5相比转化率分别提高了20%、80%和140%。同时,对FA-40进行300 h的长周期加氢实验(见图7),结果表明,这种大孔容、低堆积密度的三次纳米自组装主客体催化剂具有良好的加氢稳定性,其脱硫、脱氮活性较高,较好的芳烃饱和能力体现了其抑制结焦的性能。
3 结 论
三次纳米自组装法合成的高活性主客体催化剂具有较好的物化性能,其主体具有低堆积密度,孔分布在10~100 nm高度集中的特点,使活性金属均匀分散地负载在催化剂主体上,含量高达35.70%,催化剂呈现6.0 nm和40 nm的双峰孔结构、比表面为114 m2·g-1。加氢性能结果表明,反应趋于稳定时,FA-40的脱硫、脱氮及芳烃饱和率分别达到94.4%、95.5%和67.9%,与F-5相比转化率分别提高了的20%、80%和140%。长周期运转实验表明主客体催化剂FA-40具有较好的加氢性能。活性的提高主要源于自组装催化剂的双峰孔结构,以及更多的金属活性位,是一类有前景的处理催化裂化柴油的加氢催化剂。
[1]Song C S.Catal.Today,2003,86(1/2/3/4):211-263
[2]EPA,Control of Air Pollution from New Motor Vehicles Amendment to the Tierce Gasoline Sulfur Regulations.US Environmental Protection Agency,April 13,2001
[3]Schulz Hans,Böhringer Walter,Waller Peter,et al.Catalysis Today,1999,49(1/2/3):87-97
[4]ZHAO Yue(赵悦),YIN Chang-Long(殷长龙),CHEN Shi-An(陈世安),et al.Petroleum Processing and Petrochemicals(石油炼制与化工),2009,40(12):26-29
[5]Whitehurst D D,Takaaki Isoda,Isao Mochida.Adv.Catal.,1998,42:345-471
[6]Startsev A N.Catal.Rev.Sci.Eng.,1995,37(3):353-423
[7]ZHANG Hai-Jiao(张海礁),BIAN Chang-Bing(边昌兵),WANG Hai-Yan(王海彦),et al.Petroleum Processing and Petrochemicals(石油炼制与化工),2011,42(12):42-45
[8]GAO Xiao-Dong(高晓冬),HU Zhi-Hai(胡志海),NIE Hong(聂红),et al.Petroleum Processing and Petrochemicals(石油炼制与化工),2002,33(Z1):36-37
[9]FANG Xiang-Chen(方向晨).Guangdong Chemical Industry(广东化工),2009,36(10):97-98,113
[11]Jung Y S,Yoon W L,Lee T W,et al.Int.J.Hydrogen Energy,2010,35:11237-11244
[12]ZHANG Cheng(张成),WANG Yong-Lin(王永林),YANG Chun-Yan(杨春雁),et al.Industrial Catalysis(工业催化),2012,20(5):31-35
[13]Pizzio L R,Vázquez P G,Cáceres C V,et al.Appl.Catal.A,2003,256:125-39
[14]Tzompantzi F J,Manriquez M E,Padilla J M,et al.Catal.Today,2008,133:154-159
[15]Franklin J.Méndez,Aura Llanos,Marcel Echeverría,et al.Fuel,2013,110:249-258
[16]WU Hui(巫辉),ZHAO Xuan(赵萱),QIAN Jing(钱菁),et al.J.Wuhan Univ.of Technol.(武汉理工大学学报),2006,28(3):113-116
[17]LIU Hui-Ping(刘惠平),LU Guan-Zhong(卢冠忠).Chinese J.Inorg.Chem.(无机化学学报),2011,27(10):2045-2052
[18]REN Shi-Biao(任世彪),QIU Jin-Heng(邱金恒),WANG Chun-Yan(王春燕),et al.Chinese J.Inorg.Chem.(无机化学学报),2007,23(6):1021-1028
[19]ZHEN Xiao-Bo(甄晓波),WANG Ding-Cong(王鼎聪),DING Wei(丁巍),et al.Journal of Petrochemical Universities(石油化工高等学校学报),2013,26(1):11-16
[20]ZHANG Jin-Hui(张锦辉),WANG Ding-Cong(王鼎聪),DING Wei(丁巍),et al.Journal of Petrochemical Universities(石油化工高等学校学报),2013,26(1):17-20
[21]XU Xiao-Ming(徐晓明),WANG Ding-Cong(王鼎聪),YANG Gang(杨刚),et al.Petroleum Processing and Petrochemicals(石油炼制与化工),2011,42(7):81-84
[22]Wang D C.Sci.China Ser.B,2007,50(1):105-113
[23]Wang D C.Sci.China Ser.B,2009,52(12):2114-2124
[24]LIU Ji-Duan(刘纪端),WANG Ding-Cong(王鼎聪).China:Patent,101890383A.2010-11-24
