助剂对Ni/SiO2催化剂微观结构及二硝基甲苯催化加氢性能的影响
2014-01-02于智慧
于智慧 闫 泽 范 辉,2 李 忠*,
(1太原理工大学煤化工研究所,煤科学与技术教育部和山西省重点实验室,太原 030024)
(2赛鼎工程有限公司技术中心,太原 030024)
甲苯二胺(TDA,C6H3CH3(NH2)2)又名二氨基甲苯,是一种重要的化工原料和化学中间体,通常采用二硝基甲苯(DNT,C6H3CH3(NO2)2)在外加有机溶剂和催化剂条件下加氢合成。在二硝基甲苯催化加氢的反应中,催化剂是影响产品成本以及产品质量的关键因素。目前,液相加氢催化剂主要有负载型Pd/C和Pt/C等贵金属催化剂[1-2]和Raney-Ni催化剂[3]。负载型Pd/C和Pt/C等贵金属催化剂具有反应压力低和催化活性高的优点,但是催化剂价格昂贵且易于积碳或中毒而失活[4];Raney-Ni催化剂价格低廉但在空气中容易自燃[5]。
负载型镍基催化剂具有价格低廉和活性高等优点,故被广泛用于多种加氢反应中,如应用于CO加氢合成甲烷和甲醇[6],硝基苯加氢合成苯胺[7]以及二硝基甲苯加氢合成甲苯二胺[8]等反应。唐博合金等[9]采用化学还原法制备了负载型Ni-B催化剂,应用于裂解汽油的加氢反应中。结果表明,载体的引入不但提高了催化剂的催化性能,而且提高了催化剂的热稳定性。李贵贤等[8]以Ni/硅藻土为催化剂,考察了反应温度、反应时间和氢气压力等条件对2,4-DNT加氢的影响。结果表明,当反应温度130℃、氢气压力3.0 MPa,反应时间140 min时,二硝基甲苯的转化率达99%。虽然负载型催化剂在DNT加氢反应中表现出较好的催化加氢性能,但其反应温度和反应压力均高于工业条件。
为了进一步提高负载型镍基催化剂的催化性能,国内外对催化剂的制备工艺进行了大量的改进型研究。助剂的引入不但有助于提高镍活性中心的分散度,而且改善了Ni的化学环境和还原性质,提高了催化剂的催化性能。陶然等[10]采用等体积浸渍法制备了Ni/SiO2负载型催化剂并应用于催化m-二硝基苯液相催化剂加氢反应中,研究表明La2O3,K2O和Li2O等助剂的引入大大提高了催化剂中镍物种的分散度和还原能力。Yan等[11]报道Fe助剂能促进Ni物种的分散,提高催化剂的稳定性,而且Fe于Ni存在电子转移,有利于加氢反应。李凤梅等[12]研究发现少量Ce助剂的添加能促进了镍物种的分散和还原,使催化剂的活性比表面积增加。杨永宁等[13]发现Fe、Mo助剂的加入可促进NiO的还原,提高NiO的分散度及表面NiO的含量。闫少伟等[14]发现La助剂的加入提高了NiO的分散度,降低了NiO的还原温度,提高了催化剂的稳定性和抗积碳能力。
本文将 Ce、Zr、La、Co 和 Fe 助剂引入至 Ni/SiO2催化剂中,通过XRD,BET,H2-TPD,H2-TPR和XPS技术对催化剂进行了表征。研究了助剂对Ni/SiO2催化剂微观结构和其催化二硝基甲苯加氢性能的影响,筛选出对二硝基甲苯加氢反应催化剂催化性能具有明显促进作用的La助剂,并通过XPS探讨了添加La助剂以后Ni2p的化学环境。
1 实验部分
1.1 催化剂的制备
采用等体积浸渍法制备负载型Ni/SiO2催化剂。先将粉末SiO2载体 (Sigma/USA,比表面积198.907 m2·g-1)在烘箱中于 110℃干燥 3 h,以除去载体内吸附的水分;然后在0.2 moL·L-1硝酸镍溶液中,加入一定量的助剂可溶盐(La(NO3)3、Fe(NO3)3、CeCl3、ZrOCl2或CoCl2中一种)形成混合溶液。再将SiO2粉末加入到上述混合溶液中,在40 r·min-1搅拌条件下常温浸渍24 h后,在烘箱中于110℃干燥8 h;将干燥后得粉末研磨至80~120目后,置于马弗炉中于500℃焙烧4 h,即得不同助剂修饰的NiO-x/SiO2前驱体。将NiO-x/SiO2在550℃和90%N2~10%H2气氛下还原5 h,降至室温即得目标催化剂,并将其保存在真空袋中备用。本文将还原前后的催化剂表示为NiO-x/SiO2和Ni-x/SiO2,其中x表示助剂种类。其中催化剂中Ni金属单质的含量为28wt%,催化剂中金属助剂氧化物中助剂元素含量为3wt%。
1.2 催化剂的性能评价
将5.0 g DNT和100 mL甲醇放入烧杯在60℃水浴中直至完全溶解,将其倒入250 mL的反应釜中,同时加入0.5 g目标催化剂。在常温条件下,分别用N2和H2置换反应釜3次后,以10℃·min-1的升温速度升至110℃,用氢气充压至2.0 MPa,并在400 r·min-1搅拌下开始反应,反应过程中不断通入H2以维持釜内压力为2.0 MPa,反应持续2 h后结束。液相产物由GC-9610型气相色谱分析仪分析其组成。使用SE-30毛细管色谱柱 (φ 0.32 mm×0.33 μm×30 m),用氢火焰离子化检测器(FID)检测。载气N2流量 30 mL·min-1,H2流量 20 mL·min-1, 空气流量200 mL·min-1,柱温100℃,进样器温度230℃,检测器温度200℃,进样量1.0 μL,进样分流比为60:1,采用外标法分析产物含量。
1.3 催化剂的表征
X射线衍射(XRD)表征在日本Rigaku D/max 2500型 X射线衍射仪上进行,Cu Kα射线(λ=0.154 056 nm),扫描速度 8°·min-1,石墨单色器,靶电压和电流分别为40 kV和100 mA,步长0.01°,扫描范围5°~85°,闪烁计数器记录强度。
H2-TPR实验在美国Micromeritics公司AutochemⅡ2920型全自动程序升温化学吸附仪上进行,实验条件:称量约40 mg催化剂用量,将催化剂样品置于U型石英反应管中,流速50 mL·min-1,以10℃·min-1的升温速率升温至150℃,采用N2气氛吹恒温吹扫30 min,降温至50℃,切换VH2∶VAr=10∶90混合气体,恒速 40 mL·min-1,系统稳定后,以10℃·min-1的速率升温至600℃,氢信号用热导池检测器(TCD)检测。
H2-TPD实验在美国Micromeritics公司AutochemⅡ2920型全自动程序升温化学吸附仪上进行。将40 mg催化剂置于U型石英反应管中。先用 30 mL·min-1的N2吹扫, 以 10℃·min-1升至200℃对样品进行预处理。然后以90℃·min-1降至40℃,用10%H2-90%Ar的混合气以30 mL·min-1的速度吸附20 min,然后切换为纯Ar,待基线稳定后,以10℃·min-1升温至600℃后脱附,用TCD检测器检测H2信号。
比表面积及比孔容 (BET)在Micromeritics ASAP2000型低温N2吸附仪测定。首先对样品进行抽真空预处理,然后在液氮温度下进行氮气吸附,并通过脱附测定样品的比表面积和比孔容。
XPS谱在Kratos Axis Ultra DLD型多功能电子能谱仪上测定,其中X射线源是Al Kα(hν=1 486.6 eV),样品表面的荷电效应用C1s(284.60 eV)校正。全谱扫描的能量范围0~1 200 eV,扫描步长1 eV,通能80 eV;窄谱扫描的步长100 meV,通能40 eV,扫描次数1次,工作电压15 kV,功率150 W,分析面积为 0.7×0.3 mm2; 真空室的真空度 1.33×10-10Pa,分辨率0.2 eV。
2 结果与讨论
2.1 催化剂活性评价
二硝基甲苯催化加氢合成甲苯二胺是一个固-液-气三相反应,在反应过程中会经过一系列的化学加氢反应,包括平行反应、连串反应等多个反应过程,因此反应温度和反应压力对DNT加氢合成TDA非常重要。前期工作研究得到,随着反应温度(90~110℃)的提高,DNT的转化率逐渐增大,当反应温度高于110℃时,DNT的转化率基本没有变化,但TDA的选择性随着温度的升高(90~130℃)逐渐降低。随着反应压力(1.0~3.0 MPa)的增大,DNT的转化率和TDA的选择性先增大后趋于稳定,反应压力在2.0 MPa时,DNT的转化率和TDA的选择性达到最大。因此,本论文催化剂在反应温度为110℃,反应压力为2.0 MPa条件下,进行催化剂催化性能评价。
表1为添加不同助剂制备的Ni/SiO2催化剂的催化活性评价结果。由表可以看出,Ni负载量为28%的Ni/SiO2没有添加任何助剂时,DNT的转化率为94.0%,产品TDA的选择性为98.0%。添加不同助剂制备的Ni/SiO2催化剂对催化二硝基甲苯合成甲苯二胺的反应都有一定影响。添加Co和Ce助剂以后,DNT的转化率和TDA的选择性都降低,其中添加Ce助剂以后,DNT的转化率和TDA的选择性分别降低为91.1%和95.9%,这可能是由于具有高储氧作用的CeO2能够促进单质Ni变成Ni2+离子,不利于加氢反应[15-17]。而添加La、Fe和Zr助剂后,DNT的转化率和TDA的选择性都有所提高。添加Fe能够形成双金属NiFe合金,二者协同作用有利于提高催化剂的催化活性[18]。其中添加La助剂制备的Ni/SiO2催化剂的催化性能最好,DNT的转化率达到98.1%,TDA的选择性达到99.1%,这主要是由于La助剂能够促进NiO的分散,提高催化剂的催化活性[19-20]。

表1 助剂对Ni/SiO2催化剂催化DNT加氢性能的影响Table 1 Influence of addictives on catalytic performance of Ni/SiO2catalysts for dinitrotoluene hydrogenation
2.2 催化剂的XRD表征
图1为添加不同助剂制备的Ni/SiO2催化剂还原前的XRD谱图。由图可以看出,各催化剂的XRD衍射峰基本相似,其中2θ=22°归属于载体SiO2的特征衍射峰,2θ=37°,43°和 63°左右归属于 NiO 晶相的特征衍射峰。在所有谱图中均未检测到助剂的衍射峰,其主要原因可能是助剂含量较少,助剂在载体SiO2表面分散均匀,没有形成XRD可以检测到的晶相。与未添加助剂制备的Ni/SiO2催化剂的XRD谱图相比,添加助剂以后,催化剂中的NiO晶相的衍射峰强度都有不同程度的降低,其中添加La助剂制备的催化剂,NiO晶相的衍射峰强度最弱。通过Jade软件对各催化剂XRD谱图2θ=43°左右NiO衍射峰的半峰宽进行数据采集,然后通过谢乐公式计算了催化剂中NiO的晶粒尺寸,详见表2。由表2可以看出,未添加助剂制备的Ni/SiO2催化剂,NiO晶粒大小为21.1 nm。添加不同助剂制备的Ni/SiO2催化剂中NiO晶粒大小都有不同程度的减小,添加La助剂制备的Ni/SiO2催化剂NiO晶粒尺寸为17.6 nm,为最小。可见,La助剂能够较好的促进NiO在载体SiO2分散,从而使得NiO晶粒减小。

图1 不同助剂制备的Ni/SiO2催化剂还原前XRD谱图Fig.1 XRD patterns of Ni/SiO2catalysts with different additives before reduction
图2为添加不同助剂制备的Ni/SiO2催化剂还原后的XRD谱图。由图可以看出,各催化剂谱图中都出现了 2θ=22°的 SiO2的特征衍射峰,2θ=44°,52°和76°左右的单质Ni晶粒的特征衍射峰[21-23]。在所有谱图中均未检测到NiO晶相的衍射峰,可见在本文的还原条件下,NiO能够全部或部分被还原为Ni单质,即使存在未被还原的NiO也没有形成XRD可以检测到的晶相。与未添加助剂制备的Ni/SiO2催化剂相比,添加助剂以后,催化剂中的单质Ni晶粒衍射峰强度都有不同程度的减弱。与Co和Ce助剂相比,添加La、Fe和Zr助剂的制备的Ni/SiO2催化剂使得单质Ni衍射峰强度变的更低。其中添加La助剂制备的Ni/SiO2催化剂中单质Ni晶粒的衍射峰强度最弱。与计算还原前NiO晶粒尺寸大小方法一样,通过Jade软件对各催化剂XRD谱图中2θ=44°左右衍射峰的半峰宽进行数据采集,然后通过谢乐公式计算催化剂中Ni的晶粒尺寸,详见表2。由表2可以看出,未添加助剂制备的Ni/SiO2催化剂单质Ni晶粒大小为22.5 nm。添加不同助剂以后,催化剂中Ni晶粒都有一定程度的降低,表明助剂的添加,都能一定程度上促进Ni晶粒在载体表面的分散。与Co和Ce助剂相比,添加La、Fe和Zr助剂的催化剂使得单质Ni晶粒半径更小,其中添加La助剂制备的Ni/SiO2催化剂中单质Ni晶粒最小,可见La、Fe和Zr更有助于活性金属Ni分散,La助剂更能促进Ni晶粒在载体表面的分散。研究表明[24],La2O3在镍晶粒四周形成壁垒,在焙烧过程中抑制镍晶粒的迁移,使得Ni晶粒较小。

图2 不同助剂制备的Ni/SiO2催化剂还原后的XRD谱图Fig.2 XRD patterns of Ni/SiO2catalysts with different additives after reduction
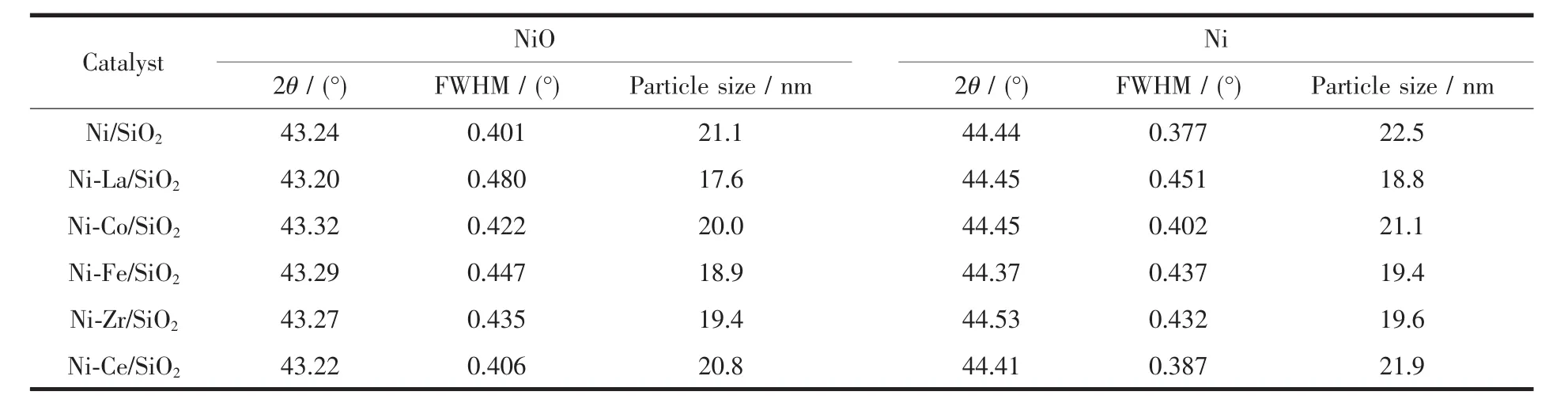
表2 不同助剂制备的NiO-x/SiO2和Ni-x/SiO2的XRD谱图分析数据Table 2 XRD analysis of NiO-x/SiO2and Ni-x/SiO2catalysts with different additives
2.3 催化剂的BET表征
表3是添加不同助剂制备的Ni/SiO2催化剂的BET表征结果。由表可见,助剂引入后催化剂的比表面积略有下降,但相差不大,表明助剂的引入导致的催化剂比表面积变化不是影响其加氢性能的主要因素。其中,负载镍催化剂Ni/SiO2与载体SiO2相比,比表面积由 199.9 m2·g-1降低到 148.9 m2·g-1,平均孔径由15.0 nm提高至29.8 nm,可见SiO2载体负载Ni后,比表面积下降了1/4,平均孔径却几乎增加一倍,这可能是由于Ni晶粒堵塞小孔所致。进一步负载助剂金属后,催化剂的比表面积、平均孔径和孔体积与Ni/SiO2相比略有下降。就催化剂的活性而言,未加入助剂制备的Ni/SiO2催化剂平均孔径为 29.8 nm,孔体积为 1.109 cm3·g-1,都为最大,但DNT的转化率和TDA的选择性分别为94.0%和98.0%,加入La助剂制备的Ni/SiO2催化剂平均孔径为26.3 nm,孔体积为0.972 cm3·g-1,DNT的转化率和TDA的选择性分别为98.1%和99.1%,加入Co助剂制备的Ni/SiO2催化剂平均孔径为24.1 nm, 孔体积为 0.875 cm3·g-1,DNT 的转化率和TDA的选择性分别为88.7%和89.3%。可见,催化剂的催化活性与平均孔径、孔体积没有相应的对应关系,本实验研究的催化剂比表面积、平均孔径和孔体积对催化活性的影响不大。
2.4 催化剂的H2-TPR表征
图3为添加不同助剂制备的Ni/SiO2催化剂的H2-TPR谱图。由图可以看出,通过分峰拟合,各催化剂均出现两个还原峰:低温还原峰PeakⅠ和高温还原峰PeakⅡ,表明催化剂存在两种不同的NiO物种,其中低温还原峰对应于均匀分散在催化剂表面的小晶粒的NiO的还原,高温峰对应于体相大颗粒NiO的还原[25]。与未添加助剂制备的Ni/SiO2催化剂相比,助剂的引入均使催化剂两还原峰温不同程度的降低。由催化剂的XRD谱图分析可知,助剂的引入导致催化剂中NiO晶粒减小,分散度增大,致使催化剂表面NiO易于还原。与其他助剂相比,添加La助剂制备的Ni/SiO2催化剂的低温还原峰温最低。

图3 不同助剂制备的Ni/SiO2催化剂的H2-TPR谱图Fig.3 H2-TPR profiles of Ni/SiO2catalysts with different additives
通过高斯拟合对添加不同助剂制备的Ni/SiO2催化剂H2-TPR谱图进行分峰处理,其分析数据结果见表4。由表可以看出,与未添加助剂制备的Ni/SiO2催化剂相比,助剂的引入不但使得低温还原峰和高温还原峰温都有所降低,而且低温还原峰的相对面积也有所增大,高温还原面积减小,表明助剂作为一种 “间隔体”,促进了NiO在载体表面的分散,增加了小颗粒NiO的量,减少了大颗粒的体相NiO,并阻止了Ni晶粒在还原过程中长大[26]。与其他助剂相比,添加La助剂以后使得NiO的低温还原峰温最低,且低温还原峰面积最大。催化剂中小晶粒NiO的含量越大,越有利于催化加氢。

表3 助剂对Ni/SiO2催化剂的孔特性的影响Table 3 Influence of additives on porosity properties of Ni/SiO2

表4 不同助剂制备的Ni/SiO2催化剂的H2-TPR分析结果Table 4 H2-TPR analytic result of Ni/SiO2with different additives
2.5 催化剂的H2-TPD表征
图4为添加不同助剂制备的Ni/SiO2催化剂的H2-TPD谱图。由图可以看出,各催化剂样品均在60℃左右出现物理吸附H2的脱附峰,同时在200~450℃出现化学吸附H2的脱附峰。与其他助剂相比,引入Fe和Ce助剂后,在200~450℃范围内出现了2个化学吸附H2的脱附峰,表明催化剂中出现了新的Ni活性中心。这可能是由于加入Fe助剂以后,Fe与Ni形成了具有加氢性能的NiFe合金,因此在334.8℃出现了吸附氢的新的Ni活性中心。有文献报道,添加Ce助剂以后,Ce会与Ni形成CeO2-Ni纳米晶粒,其归属为356.1℃出现的新的Ni活性中心[27-28]。
通过高斯拟合对添加不同助剂制备的Ni/SiO2催化剂H2-TPD谱图进行分峰处理,然后通过化学氢吸附量计算了活性比表面积,其数据结果见表5。由表可以看出,与未添加助剂制备的Ni/SiO2催化剂相比,添加助剂以后,化学吸附氢的脱附峰温均不同程度的向低温方向发生偏移,但添加Ce和Co助剂以后,催化剂的化学吸附氢量和活性比表面积均低于未添加助剂制备的Ni/SiO2。添加Zr、Fe和La助剂以后,催化剂的化学吸附氢量和活性比表面积均高于未添加助剂制备的Ni/SiO2。研究表明[29],催化剂中低吸附强度的Ni吸附中心越多,使得吸附的氢物种更易于在催化剂表面流动并参与反应,有助于催化剂的活性提高。添加La助剂以后,其化学吸附H2的脱附峰温最低,化学吸附氢量(0.291 mmol·g-1)最大,活性比表面积(8.05 m2·g-1)也最大,其催化活性最高。这是由于加入La助剂,形成了紧密的La-O-Ni结构单元,加速了H2在载体的表面的解离吸附[30],从而提高了催化活性。

图4 不同助剂制备的Ni/SiO2催化剂的H2-TPD图Fig.4 H2-TPD profiles of Ni/SiO2catalysts with different additives

表5 不同助剂制备的Ni/SiO2催化剂的H2-TPD分析数据结果Table 5 H2-TPD analytic result of Ni/SiO2catalyst samples with different additives
2.6 催化剂的XPS表征
图5为新鲜的Ni/SiO2和Ni-La/SiO2催化剂的Ni2p XPS谱图。由于La3d3/2与Ni2p3/2的图谱相重合[31-32],故进行了分峰拟合。由图可以看出,新鲜催化剂主要以Ni2+和 Ni0形式存在。Ni2+的存在,一方面是由于催化剂暴露在空气中氧化所致,另一方面其可能是在高温焙烧条件下生成Ni2SiO4[33]或La2NiO4[34],在本论文温度下不易被还原。与未添加助剂制备的Ni/SiO2催化剂相比,添加La助剂以后,Ni02p3/2的结合能由 853.03 eV降低到 852.31 eV,表明La的引入使Ni成富电子状态,致使催化剂产生更多的不饱和中心,有利于对氢物种的吸附与解离,从而提高了催化剂的催化活性[14]。

图5 新鲜的Ni/SiO2和Ni-La/SiO2催化剂的Ni2p XPS谱图Fig.5 Ni2p XPS spectra of fresh Ni/SiO2and Ni-La/SiO2 catalysts
3 结 论
在Ni/SiO2催化剂催化二硝基甲苯加氢合成甲苯二胺的的反应中,添加La、Fe和Zr助剂有助于提高催化活性,其中以La助剂的效果最佳,而添加Ce和Co降低了催化活性。La在Ni/SiO2催化剂中促进了Ni晶粒在载体SiO2表面的分散,NiO更易于还原,增加了有效活性组分Ni的数目,而且La使Ni成富电子状态,有利于氢物种的吸附与解离,提高了催化剂的催化性能,使DNT的转化率和TDA的选择性分别达到98.1%和99.1%。
[1]Neri G,Musolino M G,Milone C,et al.Appl.Catal.A:Gen.,2001,208(1/2):307-316
[2]Neri G,Rizzo G,Milone C,et al.Appl.Catal.A:Gen.,2003,249(2):303-311
[3]KONG Ling-Qi(孔令启),LI Yu-Gang(李玉刚),HAN Fang-Yu(韩方煜),et al.Chem.React.Eng.Technol.(化学反应工程与工艺),2009,25(03):244-249
[4]Zhang C,Yue H,Huang Z,et al.ACS Sust.Chem.Eng.,2012,1:161-173
[5]Hu H,Qiao M,Xie F,et al.J.Phys.Chem.,2005,109(11):5186-5192
[6]ZHANG Wei(张微),GE Qing-Jie(葛庆杰),XU Heng-Yong(徐恒泳).Chin.J.Catal.(催化学报),2010,31(11):1358-1362
[7]ZHANG Chao-Lin(张超林).Ind.Catal.(工业催化),2008,16(01):5-9
[8]LI Gui-Xian(李贵贤),YANG Lei(杨磊),MA Jian-Jun(马建军),et al.Polyurethan Ind.(聚氨酯工业),2009,24(1):30-36
[9]TANGBO He-Jin(唐博合金),LÜ Ren-Qing(吕仁庆),XIANG Shou-He(项寿鹤).Acta Petrolei Sinica:Petroleum Processing Section(石油学报:石油加工),2004,20(02):63-68
[10]TAO Ran(陶然),CHEN Ji-Xiang(陈吉祥),ZHANG Ji-Yan(张继炎).J.Chem.Ind.Eng.(化学工业与工程),2005,22(03):175-178
[11]Yan X,Sun J,Wang Y,et al.J.Mol.Catal.A:Chem.,2006,252(1/2):17-22
[12]LI Feng-Mei(李凤梅),WANG Yong-Zhao(王永钊),ZHANG Zhuo(张卓),et al.Ind.Catal.(工业催化),2011,19(11):70-74
[13]YANG Yong-Ning(杨永宁),ZHANG Huai-Ke(张怀科),LÜ En-Jing(吕恩静),et al.J.Mol.Catal.(分子催化),2011,25(1):30-36
[14]YAN Shao-Wei(闫少伟),FAN Hui(范辉),LIANG Chuan(梁川),et al.Chin.J.Catal.(催化学报),2012,33(08):1374-1382
[15]Zhu J,Peng X,Yao L,et al.Int J.Hydrogen Energy,2013,38(1):117-126
[16]Cai X,Dong X,Lin W.J.Nat.Gas Chem.,2008,17:98-102
[17]Mortola V B,Damyanova S,Zanchet D,et al.Appl.Catal.B:Environ.,2011,107:221-236
[18]LIU Hai-Bo(刘海波),CHEN Tian-Hu(陈天虎),ZHANG Xian-Long(张先龙),et al.Chin.J.Catal.(催化学报),2010,31(4):409-414
[19]Zhang R,Li F,Shi Q,et al.Appl.Catal.A:Gen.,2001,205(1/2):279-284
[20]Zhang L,Li W,Liu J,et al.Fuel,2009,88(3):511-518
[21]Wu Z,Ge S.Catal.Commun.,2011,13(1):40-43
[22]Patel N,Fernandes R,Miotello A.J.Catal.,2010,271(2):315-324
[23]Liu S,Liu Z,Wang Z,et al.Chem.Eng.J.,2008,139(1):157-164
[24]HUANG HAi-Yan(黄海燕),SHEN Zhi-Hong(沈志宏).J.University Petroleum China(石油大学学报),2000,24(3):38-41
[25]Diskin A M,Cunningham H R,Ormerod R M.Catal.Today,1998,46:147-154
[26]REN Shi-Biao(任世彪),QIU Jin-Heng(邱金恒),WANG Chun-Yan(王春燕),et al.Chinese J.Inorg.Chem.(无机化学学报),2007,23(06):1021-1028
[27]Kimura T,Miyazawa T,Nishikawa J,et al.Appl.Catal.B:Environ.,2006,68(3/4):160-170
[28]Tomishige K,Kimura T,Nishikawa J,et al.Catal.Commun.,2007,8(7):1074-1079
[29]ZHENG Yi-Xiong(郑一雄),YAO Shi-Bing(姚士冰),ZHOU Shao-Ming(周绍民).Acta Phys.-Chim.Sin.(物理化学学报),2004,20(11):1352-1356
[30]Nielsen A,Denmark H T.Catal.Rev.,1971,4(1):1-25
[31]Wojcieszak R.Appl.Catal.A:Gen.,2004,268(1/2):241-253
[32]Venezia A M,Duca D,Floriano M A,et al.Surf.Interface Anal.,1992,19:543-547
[33]Pereñíguez R,González-DelaCruz V M,Holgado J P,et al.Appl.Catal.B:Environ.,2010,93(3/4):346-353
[34]Guo J,Lou H,Zhu Y,et al.Mater.Lett.,2003,57(28):4450-4455
