Improved reversed phase liquid chromatographic method with pulsed electrochemical detection for tobramycin in bulk and pharmaceutical formulation
2013-12-23VickyMnyngEhbElkdyJosHoogmrtensErwinAdms
Vicky Mnyng, Ehb Elkdy,b, Jos Hoogmrtens, Erwin Adms,*
aLaboratory for Pharmaceutical Analysis, Faculteit Farmaceutische Wetenschappen, KU Leuven, O&N2, PB 923,Herestraat 49, B-3000 Leuven, Belgium
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Kasr El-Aini St., Cairo 11562, Egypt
1. Introduction
Tobramycin is an aminoglycoside antibiotic derived from nebramycin, an antibiotic complex produced by the fermentation of the actinomycete Streptomyces tenebrarius [1]. It is active against a broad spectrum of gram-negative bacteria and is used in a variety of pharmaceutical applications, including ophthalmic suspensions, solutions and ointments, inhalation solutions and intravenous administrations. Tobramycin can also be synthesized from kanamycin B [2]. Fig.1 shows the chemical structures of tobramycin and its major impurities.
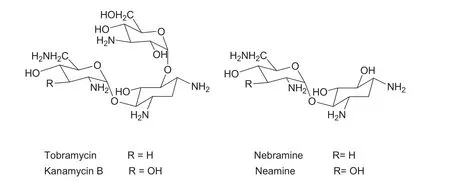
Fig.1 Chemical structures of tobramycin and its major impurities (kanamycin B, nebramine and neamine).
Tobramycin and kanamycin B are produced after base catalyzed hydrolysis of their corresponding nebramycin factors 5′-(6″-O-carbamoyltobramycin) and 4-(6″-O-carbamoyl kanamycin B) respectively, which are both produced by fermentation. Neutralization of the base with hydrochloric acid may hydrolyze tobramycin to nebramine and kanosamine, while kanamycin B may be hydrolyzed to neamine and kanosamine. Due to the complexity of the nature of the antibiotic, it is often difficult to purify the antibiotic completely. Potential impurities of tobramycin may include kanamycin B, neamine, nebramine and kanosamine. Further studies on base hydrolysis of tobramycin revealed its degradation to 2-deoxystreptamine, nebrosamine and deoxystreptaminekanosamine[3].As these related substances are often responsible for side effects, it is essential to develop a good analytical method, which will separate these impurities from the main compound, to control them.
Direct analysis of tobramycin is not simple. This is due to the polar basic nature and lack of a UV absorbing chromophore in this molecule. Several methods have been described to determine tobramycin: paper and thin-layer chromatography [4-6], gas chromatography after trimethylsilylation to increase the volatility of the analytes [7], spectrophotometry[8,9], reversed-phase LC [10-24], high performance anion exchange chromatography (HPAE) [25-27] and capillary electrophoresis[28-30].Several attempts have been made in order to enhance the detection of tobramycin.Pre-column derivatization with 2,4-dinitrofluorobenzene [10,11], o-phthalaldehyde (OPA)[12,13,19,28] and 2,4,6-trinitro-benzenesulphonic acid [16,18] as well as post-column derivatization with OPA [14,17] have been described.However,these techniques are time consuming and give problems with quantitation. Direct detection using evaporative light scattering detection (ELSD) [20], mass spectrometry (MS)[21,22], pulsed electrochemical detection (PED) [15,23-27] and capacitively coupled contactless conductivity detection (C4D)[29-30] have also been described. Due to the non linear response with ELSD, the high cost related to MS and the still poor compatibility of C4D with LC, PED is the method of choice for the detection of aminoglycoside antibiotics in combination with LC[31,32].Tobramycin can be oxidized under alkaline conditions and directly detected by PED.‘‘Pulsed''electrochemical detection is necessary to avoid fouling of the working electrode surface,what would result in a gradual decrease of the output signal.
This work describes the analysis of tobramycin and its impurities using an improved ion pair LC method with PED.The previously published method[23]which was also the basis for the actual European Pharmacopoeia (Ph. Eur.) method[33], made use of a poly(styrene-divinylbenzene) (PSDVB)column. At that time, this reversed phase polymer packing showed a better stability compared to silica based columns,but its efficiency is rather low. In the mean time, new types of silica based columns became available and column properties improved a lot. This work investigates the efficiency of such columns towards the separation of the main compound from its impurities. Compared to the previously published methods using PED[15,23-27]and the actual Ph.Eur.method[33],this method proved better performance as it is more selective and sensitive. Besides analysis of bulk samples, this method has also been used to analyze a pharmaceutical formulation containing tobramycin.
2. Experimental
2.1. Reagents and reference samples
All the reagents used were of HPLC grade. Tetrahydrofuran(THF), stabilized with 2,6-di-tert-butyl-4-methylphenol, and extra pure anhydrous sodium sulfate (ss) were obtained from Merck(Darmstadt,Germany).Potassium dihydrogen phosphate and sodium octanesulphonate (sos) were purchased from Acros Organics (Geel, Belgium). A 50% sodium hydroxide solution was from J.T. Baker (Deventer, The Netherlands); 85% phosphoric acid was from Sigma Aldrich (Steinheim, Germany).Water was produced in-house using a MilliQ water purification system(Millipore,Bedford,MA).Helium gas was obtained from Air Liquide (Li`ege, Belgium). All mobile phases were degassed by sparging helium gas.
Tobramycin samples and three known impurities (neamine,nebramine and kanamycin B) from different origins were available in our laboratory. Tobramycin Chemical Reference Substance (CRS) (91.6%) was purchased from the European Directorate for the Quality of Medicines (EDQM), Strasbourg,France. The pharmaceutical formulation Tobrex®(eye drops)manufactured by Alcon-Couvreur (Puurs, Belgium) containing 3 mg/mL tobramycin as active ingredient was purchased from a local pharmacy. Sample concentrations of 1.0 mg/mL in the mobile phase were used to investigate the related substances.The injection volume was always 10 μL.
2.2. LC instrumentation and chromatographic conditions
The LC apparatus consisted of an L-6200 Intelligent Pump(Merck Hitachi, Darmstadt, Germany), an autosampler AS 100 Spectra Series with a fixed 10 μL loop (San Jose, CA,USA), a Decade II electrochemical detector (Antec, Leyden,the Netherlands) and Chromeleon 6.70 software (Dionex Corporation, Sunnyvale, CA, USA) for data acquisition.The detector cell consisted of a gold working electrode, a HyREF reference electrode and a carbon filled polytetrafluoroethylene (PTFE) counter electrode. HyREF is an alternative for a Ag/AgCl reference electrode. It is principally maintenance free and does not contain a salt bridge.The detector cell was kept at 35°C in a hot air oven. The pulse settings were as follows; tdet400 ms with Edet+0.05 V, toxd200 ms with Eoxd+0.75 V and tred400 ms with Ered-0.15 V. Integration of the signal occurred between 200 ms and 400 ms. This triple potential-time waveform was preferred because it is applicable to different brands of PED [32]. For intermediate precision, a second detector (Decade II) was used.
During method development, the following columns were investigated: Zorbax SB C18, 250 mm×4.6 mm i.d., 5 μm(Agilent Technologies, Inc., CA, USA); Discovery C18,250 mm×4.6 mm i.d., 5 μm (Supelco, Bellefonte, PA, USA);Aqua C18, 150 mm×4.6 mm i.d., 5 μm (Phenomenex, Torrance, CA, USA); Brava BDS C18, 250 mm×4.6 mm i.d., 5 μm (Alltech, Lokeren, Belgium); and Hypurity Elite C18, 150 mm×4.6 mm i.d., 5 μm (Thermoquest, Runcorn,England). The columns were kept at a constant temperature by using a water bath with a heating immersion circulator(Julabo, Seelbach, Germany). In the final conditions, the mobile phase was composed of ss (35 g/L), sos (1 g/L), THF(14 mL/L)and 0.2 M phosphate buffer pH 3.0(50 mL/L).The 0.2 M phosphate buffer was prepared by mixing 0.2 M potassium dihydrogen phosphate with 0.2 M phosphoric acid until pH 3.0 was reached.For better detection of aminoglycosides with PED, at least pH 12 is necessary. Since the mobile phase has a lower pH, 0.5 M NaOH was pulselessly added post-column using a helium-pressurized reservoir.The column effluent was mixed with the base in a packed reaction coil from Dionex (1.2 m, 500 μL). The 0.5 M NaOH solution was prepared starting from a 50% (m/m) aqueous solution which was pipetted in helium degassed water. This solution was degassed in order to avoid the formation of carbonates that foul the electrode.
2.3. Experimental design
Factor analysis was performed by means of an experimental design and multivariate analysis using Modde 5.0 software(Umetrics, Umea, Sweden). A two level full factorial design was applied. For this design, the number of runs is equal to 2k+n, where k is the number of parameters and n is the number of center points. In this study, 4 parameters (amount of ss, amount of sos, column temperature and volume of THF) were investigated. With this number of parameters and 3 replicates of the center point,it resulted into 19 experiments.The statistical relationship between a response Y and the experimental variables Xi, Xj, … is of the following form:

where the β′s are the regression coefficients and E is the overall experimental error.
The linear coefficients, βiand βj, describe the quantitative effect of the experimental variables in the model while the cross coefficient βijmeasures the interaction effect between the variables i and j. Results are usually presented as regression coefficient plots.
3. Results and discussion
3.1. Method development
The Ph.Eur.method[33]for the determination of tobramycin was used as starting point for further improvement. This method utilizes a poly(styrene-divinylbenzene) column(PSDVB) (250 mm×4.6 mm i.d., 100 ˚A, 8 μm), maintained at 55°C. The mobile phase is prepared with carbon dioxidefree water and contains: ss (52.0 g/L), sos (1.5 g/L), THF(3.0 mL/L) and 0.2 M phosphate buffer pH 3.0 (50.0 mL/L).With this method, selectivity and sensitivity are rather poor.Hence,it was tried to improve this method by transferring it to a silica based C-18 column while preferably reducing the amount of salts in the mobile phase.
The efficiency of PSDVB as a stationary phase has as a consequence that the selectivity and the sensitivity are rather poor. On the other hand, polymer columns show a good stability and reproducibility. However,due to recent improvements in silica-based column manufacturing, their stability improved a lot. So, it was decided to adapt the Ph. Eur.method using RP C-18 columns, giving better efficiency and sensitivity so as to separate as many impurities as possible.Different silica based C-18 columns were investigated. Their selection was based on a column classification system previously developed in our laboratory [34]. The columns investigated are mentioned in Section 2.2.These columns showed a better overall selectivity. The selectivity and peak shape obtained with the Discovery column was superior to that of the other columns and this end-capped stationary phase was chosen for further investigation. The Zorbax column gave a similar selectivity,but the retention time of the main peak was about twice as high. The Aqua, Brava and Hypurity columns gave a somewhat shorter retention, but separation between peaks 10-13 was worse.
In further method development it was tried to reduce the amount of salt in the mobile phase since high salt concentrations can reduce the stability of silica-based stationary phases and may block LC pumps. Decreasing the amount of ss to 35.0 g/L leads to an increase in analysis time up to 40 min.To compensate for this, the amount of THF was increased in the mobile phase. This organic modifier also improved the selectivity, especially between kanamycin B and tobramycin.An amount of 14.0 mL/L was found to be a good compromise between the resolution and the analysis time. An optimal concentration of sos was found to be 1.0 g/L. Less than this amount leads to poor resolution of peaks while a higher amount leads to very long retention of peaks,which eventually leads to broader peak shape. The effect of temperature has been investigated between 40 and 55°C. Forty-five degree celsius gave the best overall selectivity. This temperature did not cause stability problems on a long term base. So, the final selected conditions are: mobile phase consisting of carbon dioxide-free water containing 35.0 g/L of ss, 1.0 g/L of sos,14.0 mL/L of THF and 50.0 mL/L of 0.2 M phosphate buffer pH 3.0 utilizing a Discovery C18 column kept at 45°C.A typical chromatogram obtained under the final selected chromatographic conditions for an impure sample is shown in Fig.2.As can be seen,neamine,nebramine,kanamycin B and 15 unknown impurities are well separated from the main peak.Compared to our previously published method where a total of 8 peaks were separated [23] and which served as basis for the Ph.Eur.Method[33],this new method shows better selectivity.Generally, ion pair LC shows better selectivity of peaks compared to the published HPAE methods[21,26].Hence,it is a reliable approach for impurity determination of aminoglycoside antibiotics.
3.2. Method validation
3.2.1. Factorial analysis
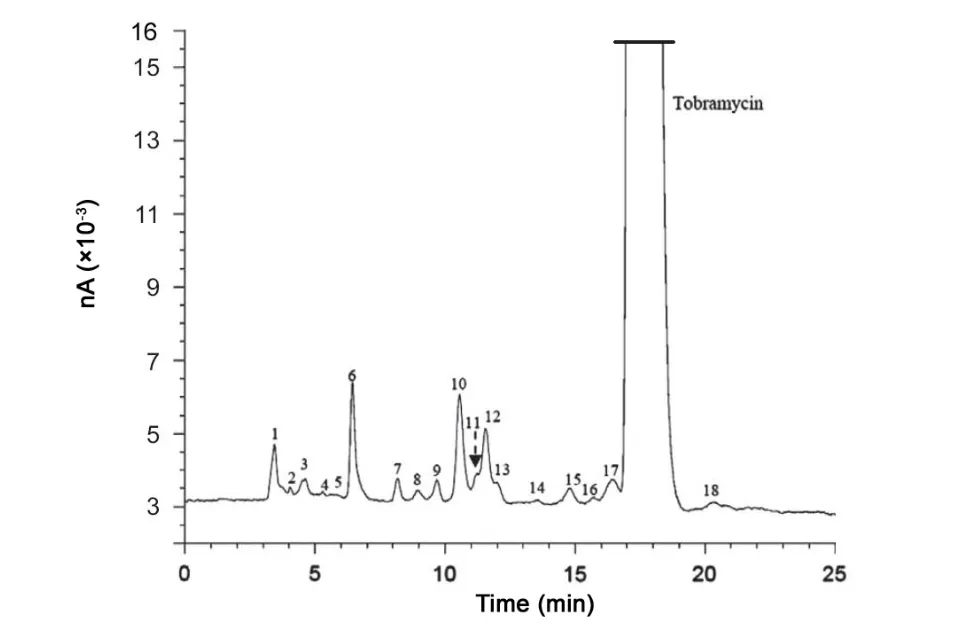
Fig.2 Typical chromatogram obtained after analyzing a tobramycin bulk sample using the improved method(nA:nano Amperes).The known impurity peaks are 7=neamine, 9=nebramine and 15=kanamycin B. The remaining impurities are of unknown identity.
Factorial analysis was performed by means of an experimental design as mentioned in Section 2.3. The different chromatographic parameter settings in the design are given in Table 1.A tobramycin bulk sample spiked with known impurities(neamine, nebramine and kanamycin B) was used for this study. The responses investigated included resolutions between:peak 5 and peak 6 (Rs1), neamine and nebramine (Rs2),nebramine and peak 10 (Rs3), peak 10 and peak 12 (Rs4) and kanamycin B and tobramycin (Rs5). The results expressed as regression coefficient plots are summarized in Fig.3.These plots consist of bars, which correspond to the regression coefficients.The bars denoted by one variable reflect the regression coefficient for the linear effect of that particular variable and the bars denoted by variable1*variable2 reflect the interaction between the two variables concerned. The 95% confidence interval is expressed in terms of an error line over the coefficient.When the interval includes zero, the variation of the response caused by changing the variable is smaller than the experimental error and the effect is considered to be not significant. The magnitude of an effect is proportional to its respective regression coefficient(See Eq. (1)). A positive regression coefficient stands for a positive effect on the resolutions studied, while a negative regression coefficient indicates a negative effect. It can be observed that ss was found to have a negative, significant effect on Rs1,Rs3 and Rs4.This means that the resolution between the peak pairs involved will decrease with an increase in the amount of ss.A similar effect is also observed with THF on Rs3 and Rs4 and with the column temperature on Rs4 and Rs5. Sos is observed to have a negative, significant effect on Rs3 while a positive, significant effect is observed on Rs4. No other significant effects were observed. Significant positive interactions were observed between sos and column temperature for Rs2 and Rs3, and between THF and column temperature for Rs2 and Rs5. Other interactions were found to be not significant. Rs5 is used as a system suitability test in the Ph. Eur. [33] for the analysis of tobramycin.For this resolution,a positive interaction was observed between THF and temperature. In order to better estimate the influence of these parameters on Rs5, a response surface plot was constructed.Fig.4 shows the variation of Rs5 as a function of THF and temperature while the other parameters are kept constant at their central values. It is observed that a change of these parameters within the range examined will not be problematic since the minimum resolution is 4.4.In the Ph.Eur.the limit is 3.
3.2.2. Quantitative aspects
The sensitivity, precision and linearity of the method were evaluated using a commercial tobramycin bulk sample. The limit of detection (LOD) (corresponding to a signal-to-noise ratio of 3) and limit of quantitation (LOQ) (corresponding to a signal-to-noise ratio of 10) were assessed. The LOD was found to be 1.7 ng, corresponding to 0.02%, and the LOQ 5 ng, corresponding to 0.05% of the total tobramycin injected(100%=1.0 mg/mL). With the polymer column, LOQ values were determined using tobramycin's main impurities and they were found to be 0.06%for neamine,0.1%for nebramine and 0.15% for kanamycin B [25]. The polymer based method was linear up to 12%. With this new method, the linearity was checked for the area of the tobramycin peak over the concentration range LOQ-12%. However, the response of the detector in this range was not linear. Due to the more narrow peaks compared to the polymer based method,overloading of the detector was clearly noticed for concentrations above 6%. This implies that a lower concentration is required for the assay of tobramycin. Upon further study,good linearity (y=1374.5x+383.5, where: y=peak area and x=concentration %) was obtained from LOQ to 6%, with a coefficient of determination (R2)=0.998.

Table 1 Chromatographic parameter settings applied in the factorial analysis, corresponding to low (-), central (0) and high (+) levels.

Fig.3 Regression coefficient plots obtained from the factorial analysis. Rs1=resolution between peak 5 and peak 6, Rs2=resolution between neamine and nebramine, Rs3=resolution between nebramine and peak 10, Rs4=resolution between peak 10 and peak 12 and Rs5=resolution between kanamycin B and tobramycin THF=volume of tetrahydrofuran, temp=column temperature,sos=amount of sodium octanesulphonate,ss=amount of sodium sulfate.

Fig.4 Response surface plot:the influence of temperature(temp)and tetrahydrofuran (THF) on the separation of kanamycin B and tobramycin (Rs5). The other parameters are kept at their central points.
To evaluate the precision of the method, the repeatability and intermediate precision on two days were checked at a concentration of 0.05 mg/mL(5%).On the different days,two different detectors and independently prepared solutions were examined. Tobramycin CRS (91.6%) was used to determine the percent content of the main compound. Tobramycin bulk sample and tobramycin CRS were each injected six times(n=6) in each detector. Expressed as relative standard deviation (RSD), the intraday repeatability amounted 0.3% (n=6)and the intermediate precision 0.6% (n=12). These results demonstrate the good precision of the method.
The accuracy was evaluated by calculating the recoveries of a 0.04,0.05 and 0.06 mg/mL tobramycin solution spiked to an equal volume of 0.05 mg/mL Tobrex® solution. They were found to be 99.4%, 98.7% and 101.1% respectively.
3.2.3. Analysis of commercial bulk samples
The developed LC-PED method was applied to the analysis of commercial samples of tobramycin. Four different samples were dissolved and diluted with the mobile phase to the appropriate concentrations. The impurity profile was investigated at a concentration of 1.0 mg/mL. Data obtained are summarized in Table 2. All related components are expressed as tobramycin, using chromatograms obtained with a 1%dilution of the sample as reference.
3.2.4. Analysis of a pharmaceutical formulation containing tobramycin
Ophthalmic preparations containing tobramycin are widely used pharmaceuticals. One such preparation (Tobrex®) was analyzed using the developed LC-PED method. To determine the assay value of tobramycin from the eye drops, samples were diluted to a concentration of 0.05 mg/mL. The content was determined using tobramycin CRS (91.6%). Typical chromatograms of the formulation and tobramycin CRS areshown in Fig.5. As can be seen from these chromatograms,there is no interference of excipients from the formulation analyzed.The average content was found to be 103.9%(n=6,RSD=1%).

Table 2 Analysis of commercial tobramycin bulk samples (%).
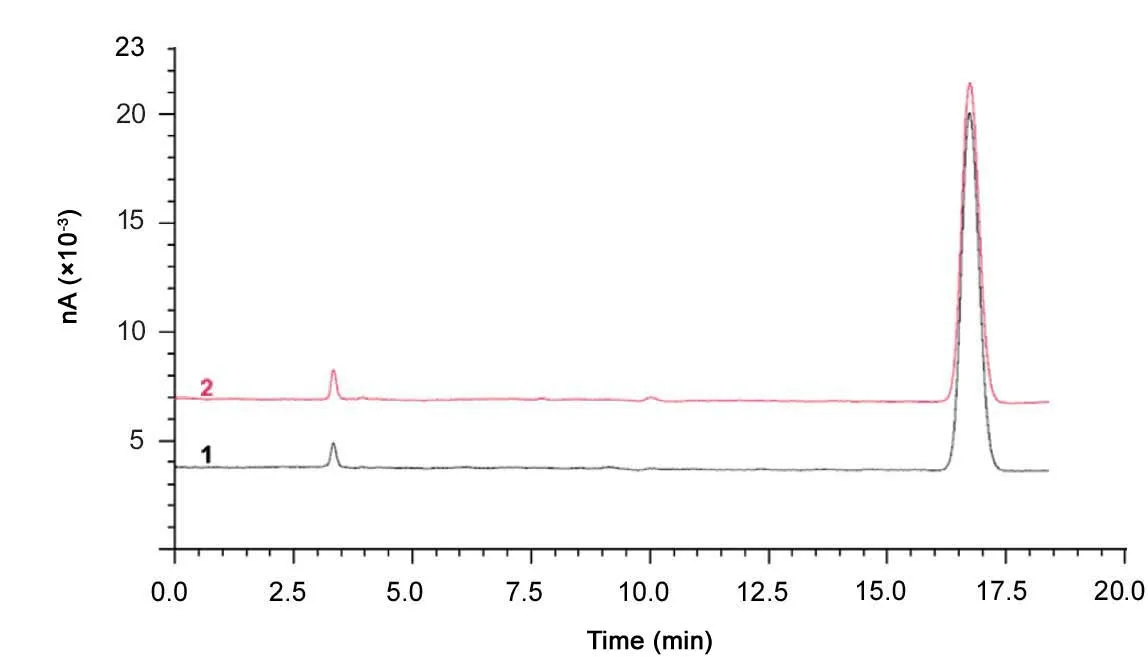
Fig.5 Typical chromatograms obtained after analyzing(1) Tobrex® and (2) tobramycin CRS using the improved LCPED method. Sample concentration was 0.05 mg/mL.
4. Conclusion
An improved LC-PED method has been developed for the analysis of tobramycin. This method allows better separation of tobramycin from its related substances, compared to existing methods. The method allows good separation of tobramycin from 17 impurities. The selectivity of the method was found to be better than other methods already published.PED has the advantage that it is a direct detection method allowing to obtain sensitive and repeatable results. The method has been used to analyze some commercial bulk samples and a pharmaceutical preparation. This method can be a suitable alternative for the current LC method in the monograph of tobramycin in the Ph. Eur.
[1] C.E.Higgins,R.E.Kastner.,Nebramycin,a new broad-spectrum antibiotic complex. II. Description of Streptomyces tenebrarius,Antimicrob. Ag. Chemother. 7 (1967) 324-331.
[2] Y. Takegi, T. Tsuchiya, S. Umezaya, et al., Synthesis of 3-deoxykanamycin B, J. Antibiot. 26 (1973) 403-406.
[3] M. Brandl, L. Gu, Degradation of tobramycin in aqueous solution, Drug Dev. Ind. Pharm. 18 (1992) 1423-1436.
[4] R.L. Hussey, Paper chromatography of tobramycin and some related compounds, J. Chromatogr. 92 (1974) 457-460.
[5] J.K`ad`ar-Pauncz,Separation of nebramycin components by thinlayer chromatography, J. Chromatogr. 170 (1979) 203-207.
[6] G. Eneva, B. Nikolova-Damyaniva, S. Spassov, et al., Determination of nebramycin components by thin-layer chromatography and densitometry, J. Planar Chromatogr. 3 (1990) 232-235.
[7] J.W. Mayhew, S.L. Gorbach, Gas-liquid chromatographic method for the assay of aminoglycoside antibiotics in serum,J. Chromatogr. 151 (1978) 133-146.
[8] J.A. Ryan, Colorimetric determination of gentamicin, kanamycin, tobramycin, and amikacin aminoglycosides with 2,4-dinitrofluorobenzene, J. Pharm. Sci. 73 (1984) 1301-1302.
[9] S.S. Sampath, D.H. Robinson, Comparison of new and existing spectrophotometric methods for the analysis of tobramycin and other aminoglycosides, J. Pharm. Sci. 79 (1990) 428-431.
[10] H. Russ, D. McCleary, R. Katimy, et al., Development and validation of a stability-indicating HPLC method for the determination of tobramycin and its related substances in an ophthalmic suspension, J. Liq. Chrom. Rel. Technol. 21 (1998)2165-2181.
[11] D.M. Barends, C.L. Zwaan, A. Hulshoff, Micro-determination of tobramycin in serum by high-performance liquid chromatography with ultraviolet detection, J. Chromatogr. 225 (1981) 417-426.
[12] J. Marples, M.D.G. Oates., Serum gentamicin, netilmicin and tobramycin assays by high performance liquid chromatography,J. Antimicrob. Chemother. 10 (1982) 311-318.
[13] L. Essers, An automated high-performance liquid chromatographic method for the determination of aminoglycosides in serum using pre-column sample clean-up and derivatization,J. Chromatogr. 305 (1984) 345-352.
[14] H. Kubo, T. Kinoshita, Y. Kobayashi, et al., Micro-scale method for determination of tobramycin in serum using high-performance liquid chromatography, J. Liq. Chromatogr. 7 (1984) 2219-2228.
[15] J.A. Polta, D.C. Johnson, K.E. Merkel, Liquid chromatographic separation of aminoglycosides with pulsed amperometric detection, J. Chromatogr. 324 (1985) 407-414.
[16] P. Gambardella, R. Punziano, M. Gionti, et al., Quantitative determination and separation of analogues of aminoglycoside antiobiotcs by high-performance liquid chromatography, J. Chromatogr.348 (1985) 229-240.
[17] H. Fabre, M. Sekkat, M.D. Blanchin, et al., Determination of aminoglycosides in pharmaceutical formulations—II. Highperformance liquid chromatography, J. Pharm. Biomed. Anal.7 (1989) 1711-1718.
[18] A.K. Dash, R. Suryanarayanan, A liquid chromatographic method for the determination of tobramycin, J. Pharm. Biomed.Anal. 9 (1991) 237-245.
[19] F. Lai, T. Sheenan, Enhancement of detection sensitivity and cleanup selectivity for tobramycin through pre-column derivatization, J. Chromatogr. 609 (1992) 173-179.
[20] N.C. Megoulas,M.A Koupparis,Development and validation of a novel HPLC/ELSD method for the direct determination of tobramycin in pharmaceuticals,plasma and urine,Anal.Bioanal.Chem. 382 (2005) 290-296.
[21] M.X. Guo, L. Wrisley, E. Maygoo, Measurement of tobramycin by reversed-phase high-performance liquid chromatography with mass spectrometry detection, Anal. Chim. Acta 571 (2006)12-16.
[22] B.Li,A.Van Schepdael,J.Hoogmartens,et al.,Characterization of impurities in tobramycin by liquid chromatography-mass spectrometry, J. Chromatogr. A 1216 (2009) 3941-3945.
[23] J. Szunyog, E. Adams, E. Roets, et al., Analysis of tobramycin by liquid chromatography with pulsed electrochemical detection,J. Pharm. Biomed. Anal. 23 (2000) 891-896.
[24] C. Ghinami, V. Giuliani, A. Menarini, et al., Electrochemical detection of tobramycin or gentamicin according to the European Pharmacopoeia analytical method,J.Chromatogr.A 1139(2007)53-56.
[25] J.A. Statler, Determination of tobramycin using high-performance liquid chromatography with pulsed amperometric detection,J. Chromatogr. 527 (1990) 244-246.
[26] V.P. Hanko, J.S. Rohrer, H.H. Liu, et al., Identification of tobramycin impurities for quality control process monitoring using high-performance anion-exchange chromatography with integrated pulsed amperometric detection, J. Pharm. Biomed.Anal. 47 (2008) 828-833.
[27] V.P. Hanko, J.S. Rohrer, Determination of tobramycin and impurities using high-performance anion exchange chromatography with integrated pulsed amperometric detection, J. Pharm. Biomed.Anal. 40 (2006) 1006-1012.
[28] E. Kaale, A. Van Schepdael, E. Roets, et al., Development and validation of capillary electrophoresis method for tobramycin with precapillary derivatization and UV detection, Electrophoresis 23 (2002) 1695-1701.
[29] W.S. Law, P. Kuban, L.L. Yuan, et al., Determination of tobramycin in human serum by capillary electrophoresis with contactless conductivity detection, Electrophoresis 27 (2006)1932-1938.
[30] M.N. El-Attug, J. Hoogmartens, E. Adams, et al., Optimization of capillary electrophoresis method with contactless conductivity detection for the analysis of tobramycin and its related substances, J. Pharm. Biomed. Anal. 58 (2012) 49-57.
[31] E. Adams, J. Hoogmartens., The application of pulsed electrochemistry to the detection of aminoglycoside antibiotics in liquid chromatography, Curr. Top. Electrochem. 10 (2004) 63-70.
[32] S. Chopra, G. Vanderheyden, J. Hoogmartens, et al., Comparative study on the analytical performance of different detectors for the liquid chromatographic analysis of tobramycin, J. Pharm.Biomed. Anal. 53 (2010) 151-157.
[33] European Pharmacopoeia, The Council of Europe, 7th ed.,Strasbourg Cedex, France, 2011, pp. 3103-3104.
[34] Column Classification System. A Roadmap for Column Users〈http://pharm.kuleuven.be/pharmchem/columnclassification.htm〉.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Cover Figure
- Fluorescence spectroscopy of osthole binding to human serum albumin
- Simultaneous quantification of prodrug oseltamivir and its metabolite oseltamivir carboxylate in human plasma by LC-MS/MS to support a bioequivalence study
- Release test of alliin/alliinase double-layer tablet by HPLC—Allicin determination
- Accurate quantitation standards of glutathione via traceable sulfur measurement by inductively coupled plasma optical emission spectrometry and ion chromatography
- Determination and stress studies on YK-1101,a potential histone deacetylase, by HPLC-UV and HPLC-TOF/MS methods