Simultaneous quantification of prodrug oseltamivir and its metabolite oseltamivir carboxylate in human plasma by LC-MS/MS to support a bioequivalence study
2013-12-23AjayGuptaSwatiGuttikarPranavShrivastavMallikaSanyal
Ajay Gupta, Swati Guttikar, Pranav S. Shrivastav, Mallika Sanyal*
aChemistry Department, Kadi Sarva Vishwavidyalaya, Sarva Vidyalaya Campus, Sector 15/23, Gandhinagar 382015, India
bBioanalytical Research Department, Veeda Clinical Research, Ambawadi, Ahmedabad 380015, India
cDepartment of Chemistry, School of Sciences, Gujarat University, Ahmedabad 380009, India
dChemistry Department, St. Xavier's College, Navrangpura, Ahmedabad 380009, India
1. Introduction
Oseltamivir (OST) is a potent neuraminidase inhibitor that is effective against both influenza A and influenza B. It selectively inhibits the neuraminidase enzymes that are essential for the release of progeny influenza viruses from host cells, and thus prevents infection of new host cells and viral spread throughout the respiratory tract [1,2]. OST (as oseltamivir phosphate, Tamiflu®) is an ethyl ester prodrug which is readily absorbed from the gastrointestinal tract with an absolute bioavailability of 75-80%. It is rapidly hydrolyzed in vivo by hepatic carboxyesterases to its active metabolite oseltamivir carboxylate (OSTC) [3]. Following oral administration of OST in healthy volunteers, oseltamivir carboxylate is detectable in plasma in about 30 min, while the maximum plasma levels (Cmax) are attained within 5 h [4]. OST is eliminated primarily by renal excretion of the active metabolite(63% as OSTC and 3% of 100 mg dose as unchanged drug).OSTC is only about 43%protein bound and has a relatively long elimination half-life of 6-10 h. Further, variations in OST pharmacokinetic parameters in patients with mild to moderate hepatic or renal impairment are not clinically significant [5,6].
Several assays have been reported for the determination of oseltamivir and/or its active metabolite oseltamivir carboxylate in different biological matrices such as rat plasma[7-10],mouse plasma[9],rat urine[9],rat cerebrospinal fluid[10],rat brain [10], rat dried blood spot (DBS) [11], human DBS [11],human saliva[12],human urine[9,10,12,13],human blood[13],human tissue [13], human serum [14] and human plasma[9,10,12,15-18]. Although there are many reports dealing with simultaneous estimation of OST and OSTC in biological samples [8-12,16,18], relatively few methods have been used for bioequivalence/pharmacokinetic study in patients [18] or healthy volunteers [4,5,14]. Moreover, there are no reports on such studies done with Indian subjects. Schentag and co-workers [4] have studied pharmacokinetics of OST and OSTC in healthy Japanese and Caucasian subjects with 75 and 150 mg OST doses. The study showed similar pharmacokinetics in both type of subjects. In another report, the pharmacokinetics and tolerability of oral OST was investigated in healthy and elderly subjects [5]. Bahrami et al. [14]have determined OSTC in human serum by HPLC-UV and applied the method for a cross-over bioequivalence study of two OST preparations in 24 healthy volunteers. Kanneti et al.[16]proposed a highly rapid method for determination of OST and OSTC in spiked human plasma; however, the pharmacokinetic data were not presented.Very recently,Kromdijk et al.[18] described a method for determination of OST and OSTC in human fluoride EDTA plasma by LC-ESI-MS/MS with a sensitivity of 3.0 and 10 ng/mL respectively employing 50 μL plasma samples. The method was used to determine both the analytes in specific patient populations to evaluate current dosing regimen. A comparative assessment of different methods developed for OST and/or OSTC in rat and human plasma is presented in Table 1.
Development of reliable, rugged and sensitive methods for the simultaneous determination of OST and OSTC is essential as the ex vivo stability of OST is a major concern due to its rapid conversion to OSTC by plasma esterase enzymes,which can lead to pharmacokinetic variations [18]. Further,to establish suitable dosing regimens it is indispensable to consider different populations for pharmacokinetic/bioequivalence applications. Thus, in the present work a highly sensitive and selective LC-MS/MS method has been developed for measuring plasma concentration of OST and OSTC.The method offers reduced overall analysis time (extraction and chromatography) with minimum matrix interference and requires low amounts of toxic organic solvents for sample analysis.The wide linear dynamic concentration range ensures estimation of the analytes with desired accuracy and precision in human volunteers for a bioequivalence study. Further, the assay reproducibility is successfully demonstrated by reanalysis of 151 subject samples.
2. Experimental
2.1. Chemicals and materials
tandards of OST (purity, 99.9%), OSTC (purity,99.3%), oseltamivir-d5 (IS-1, purity, 100%) and oseltamivir carboxylate-C13-d3 (IS-2, purity, 98.0%) were procured from Hetero Drugs Limited (Hyderabad, India), Neucon Pharma Pvt. Ltd. (Goa, India), Vivan Life Sciences Pvt. Ltd. (Mumbai, India) and Clearsynth Labs Pvt. Ltd. (Mumbai, India)respectively. HPLC grade methanol, acetonitrile, ammonium formate and formic acid were obtained from Merck Specialties Pvt. Ltd. (Mumbai, India). Dichlorvos was obtained from Sigma Aldrich Chemicals Pvt.Ltd.(Bangalore,India).Orochem DVB-LP (30 mg, 1 cc) cartridges were obtained from Orochem Technologies Inc. (Illinois, USA). Water used in the entire analysis was prepared using Milli-Q water purification system from Millipore (Bangalore, India). Blank human plasma was obtained from Supratech Micropath (Ahmedabad, India) and was stored at -20°C until use.
2.2. Liquid chromatography and mass spectrometric conditions
?
A Shimadzu LC-VP HPLC system (Kyoto, Japan) was used for chromatographic separation of OST,OSTC,IS-1 and IS-2 on a Symmetry C18 (100 mm×4.6 mm, 5 μm) analytical column,maintained at 40°C in the column oven.For isocratic elution, the mobile phase consisting of 10 mM ammonium formate and acetonitrile (30:70, v/v) was delivered at a flowrate of 1.0 mL/min.The total eluate from the column was split in 70:30 (v/v) ratio; flow directed to the electrospray interface was equivalent to 300 μL/min. The autosampler temperature was maintained at 5°C and the average pressure of the system was 1500 psi. A triple quadrupole mass spectrometer, MDS SCIEX API-4000 (Toronto, Canada), equipped with electro spray ionization and operating in positive ionization mode was used for detection of analytes and ISs. For quantitation,multiple reaction monitoring (MRM) was used to monitor precursor→product ion transitions at m/z 313.1→166.2,285.1→138.1, 318.1→171.2 and 289.2→138.3 for OST,OSTC, IS-1 and IS-2 respectively. The source dependent parameters set, nitrogen (purity, 99.95%) as Gas 1 (nebulizer gas) and Gas 2 (heater gas), ion spray voltage, heater temperature, curtain gas nitrogen and collisional activation dissociation were optimized at 40 psig and 60 psig, 4000 V,400 °C, 10 psig and 3 psig respectively. Other compound dependent parameters like declustering potential, entrance potential, collision energy and exit cell potential were maintained at 15.0, 10.0, 25.0 and 10.0 V respectively for both the analytes and ISs, while the dwell time was set at 200 ms. Data collection, peak integration, and calculations were performed using the Analyst software version 1.4.2.
2.3. Calibration standards and quality control samples
Stock solutions of OST (1000 μg/mL) and OSTC (1000 μg/mL) were prepared by dissolving accurately weighed reference standards in water. Mixed stock solution of OST (100 μg/mL)and OSTC (400 μg/mL) was prepared by taking 200 μL of OST and 800 μL of OSTC stock solution and made up to 2.0 mL with dichlorvos solution (4 mg/mL in acetonitrile ).Working solutions of the analytes were prepared by serial dilution of mixed stock solution in dichlorvos solution.Calibration standards (CSs) and quality control (QC) samples were prepared by spiking blank plasma(2%of total volume of blank plasma) with mixed stock solutions. CSs were made at 0.5, 1.0, 4.0, 10.0, 20.0, 40.0, 100 and 200 ng/mL concentrations for OST and 2.0, 4.0, 16.0, 40.0, 80.0, 160, 400 and 800 ng/mL concentrations for OSTC. QC samples were prepared at four concentration levels,160/640 ng/mL(HQC,high quality control), 80.0/320 ng/mL (MQC, medium quality control), 1.5/6.0 ng/mL (LQC, low quality control) and 0.5/2.0 ng/mL (LLOQ QC, lower limit of quantification quality control) for OST/OSTC respectively. Stock solutions of IS-1 and IS-2 (200 μg/mL each) were prepared by dissolving requisite amount in water. An aliquot of 25 μL of IS-1(200 μg/mL) and 500 μL of IS-2 (200 μg/mL) was further diluted to 100 mL with dichlorvos solution to obtain a solution of 50 ng/mL for IS-1 and 1000 ng/mL for IS-2. All the solutions (standard stock, CSs and QC samples) were stored at 5°C until use.
2.4. Sample extraction procedure
Prior to analysis, all frozen subject samples, CSs and QC samples were thawed in ice bath maintained below 10°C. To an aliquot of 200 μL of spiked plasma sample, 50 μL of mixed internal standard was added and vortexed for 15 s.Further, 500 μL of 1.0% formic acid in water was added and vortexed for another 15 s. Samples were then centrifuged at 3204×g for 2 min at 10°C and loaded on Orochem DVB-LP (1 cc, 30 mg) extraction cartridges which were preconditioned with 1 mL of methanol followed by 1 mL of water. The cartridges were washed twice with 1% formic acid in water. The analytes and ISs were eluted with 0.2 mL of dichlorvos solution (0.1 mg/mL in acetonitrile): water(70:30, v/v). Samples were transferred to pre-labeled autosampler vial and 10 μL was used for injection in the chromatographic system.
2.5. Procedures for method validation
The method was validated as per the USFDA guidelines[19,20]. System suitability experiment was performed by injecting six consecutive injections using aqueous standard mixture of OST (80 ng/mL), OSTC (320 ng/mL), IS-1 (500 ng/mL) and IS-2 (1000 ng/mL) at the start of each batch during method validation.System performance was studied by injecting one extracted LLOQ sample with IS and the autosampler carryover of analytes was experimentally determined by sequentially injecting extracted blank plasma→upper limit of quantitation (ULOQ) sample→extracted blank plasma→LLOQ sample→extracted blank plasma at the beginning of each analytical batch.
The selectivity of the method was evaluated by analyzing ten different batches of plasma, which included seven K3EDTA and one each of lipidemic, haemolysed and heparinised plasma. Interference of commonly used medications by human volunteers was checked for acetaminophen, cetirizine,domperidone, ranitidine, diclofenac, ibuprofen, nicotine and caffeine in six different batches of plasma having K3EDTA as an anticoagulant. Their stock solutions were prepared by dissolving requisite amount in methanol and water (50:50,v/v). Further, a mixed working solution of acetaminophen(1000 μg/mL), cetirizine (20 μg/mL), domperidone (1 μg/mL), ranitidine (27.5 μg/mL), diclofenac (100 μg/mL), ibuprofen (2250 μg/mL), nicotine (5 μg/mL) and caffeine(1000 μg/mL) was prepared in the same diluents, spiked in plasma and analyzed under the same conditions at LQC and HQC levels in six replicates. These sets were processed along with freshly prepared CSs and qualifying QC samples in duplicate.
The linearity of the method was ascertained by measuring the peak area ratio response (analyte/IS) for five calibration curves containing eight non-zero concentrations. Each calibration curve was analyzed individually by using least square weighted (1/x2) linear regression.
Intra-batch accuracy and precision (% CV) were determined in six replicates of QC samples along with CSs. The inter-batch accuracy and precision were assessed by analyzing five precision and accuracy batches on three consecutive days.Reinjection reproducibility was also checked by re-injecting one entire validation batch.
The extraction recovery for analytes and ISs was calculated by comparing the mean area response of extracted samples(spiked before extraction) with that of unextracted samples(spiked after extraction) at HQC, MQC and LQC levels.Matrix effect (expressed as internal standard normalized matrix factor) was assessed by comparing the mean area response of unextracted samples (spiked after extraction) with mean area of neat standard solutions at three QC levels.Qualitative illustration of matrix ion suppression/enhancement was conducted by post column infusion of analytes(MQC level) at 10 μL/min by a Harvard infusion pump through a ‘T' connector [21].
Stability was examined by measuring the area ratio response(analyte/IS) of stability samples against freshly prepared comparison standards at LQC and HQC levels. Stock and working solutions of analytes and mixed ISs solutions were checked for short term stability at room temperature and long term stability at 5°C.Autosampler stability(wet extract),bench top(at room temperature)stability and freeze-thaw stability were determined at LQC and HQC using six replicates at each level. Long term stability of spiked plasma samples stored at-20°C and-70°C was also studied at both these levels.
Method ruggedness was evaluated on two precision and accuracy batches. The first batch was analyzed by different analysts while the second batch was studied on two different equipments and columns of the same make. Dilution reliability was determined by diluting the stock solution prepared as spiked standard at 300 ng/mL for OST and 1200 ng/mL for OSTC in the screened plasma. The precision and accuracy for dilution integrity standards at 1/2 and 1/10th dilution were determined by analyzing the samples against freshly prepared CSs.
2.6. Application of the method in healthy subjects and incurred sample reanalysis
The validated method was applied to quantify plasma OST and OSTC concentration for a bioequivalence study in 42 healthy Indian subjects after oral administration of test(75 mg capsules from an Indian Company) and a reference (TAMIFLU®,75 mg oseltamivir phosphate capsules from Genentech USA Inc., USA) formulation under fed conditions. Written consent was taken from all the subjects after informing them about the objectives and possible risks involved in the study.The study was conducted strictly in accordance with the guidelines laid down by International Conference on Harmonization and USFDA[22].Blood samples were collected at 0.0(pre-dose), 0.25, 0.50, 0.75, 1.00, 1.50, 2.00, 2.50, 3.00, 3.50,4.00, 4.50, 5.00, 5.50, 6.00, 6.50, 7.0, 8.0, 9.0, 10.0, 12.0, 24.0,36.0 and 48.0 h after oral administration of test and reference formulation in labeled K3EDTA-vacutainers placed in an ice bath maintained below 10°C. Plasma was separated through centrifugation and transferred to polypropylene tube containing 4 mg/mL of dichlorvos stock solution (plasma to dichlorvos stock solution ratio was 95:5, v/v). The samples were kept frozen at -20°C till the completion of both the periods and then below -70°C until analysis. The pharmacokinetic parameters for OST and OSTC were estimated by noncompartmental model using WinNonlin software version 5.3(Pharsight Corporation, Sunnyvale, CA, USA). An incurred sample re-analysis (ISR) was also conducted by selection of 151 subject samples near Cmaxand in the elimination phase of the pharmacokinetic profiles. As per the acceptance criterion the percent change in the values should not be more than±20% [23].
3. Results and discussion
3.1. Mass spectrometry
The present study was conducted using electrospray ionization(ESI) in the positive mode as OST, OSTC, IS-1 and IS-2 have primary and secondary amino groups. Initially, the precursor and product ions were optimized by infusing 500 ng/mL solutions in the mass spectrometer between m/z 50 and 360 range in the positive as well as negative modes. However,it was difficult to get the deprotonated precursor ion for OST in the negative mode and hence positive ionization mode was selected.Further,the use of 0.1% formic acid in the mobile phase improved the response of protonated precursor [M+H]+ions at m/z 313.1,285.1, 318.1 and 289.2 for OST, OSTC, IS-1 and IS-2 respectively in the full scan mass spectra as observed previously [18].Most intense and consistent product ions for OST, OSTC, IS-1 and IS-2 were found at m/z 166.2, 138.1, 171.2 and 138.4 respectively by applying 25 eV collision energy(Fig.1).The ions at m/z 166.2 and 138.1 for OST and OSTC respectively can be attributed to the loss of formamide and pentyloxy groups from the precursor ions. The MRM parameters like nebulizer gas,heater gas flow, ion spray voltage and source temperature were suitably optimized to obtain a consistent and adequate response for the analyte. A dwell time of 200 ms for OST, OSTC, IS-1 and IS-2 was adequate and no cross talk was observed between their MRMs.
3.2. Optimization of extraction procedure
Due to rapid hydrolysis of OST into its active metabolite OSTC it is essential to use an esterase enzyme inhibitor for their simultaneous determination, especially during a clinical study[24]. Wiltshire et al. [9] carried out extensive stability study for OST in dichlorvos(an esterase inhibitor)treated plasma samples from humans, rat, mouse, marmoset, rabbit and ferret. Similarly, Lindeg ˚ardh and co-workers [12] have successfully used fluoride/oxalate to stabilize OST in human plasma, urine and saliva.Furthermore,Chang et al.[8]have shown that hydrolysis can also be controlled by keeping the samples on ice and that there is no change in OST concentration up to 1 h. Based on these observations, the working solutions of OST and OSTC used for spiking blank plasma were prepared in dichlorvos solution (4 mg/mL in acetonitrile), while plasma obtained from subject samples was directly collected in tubes containing dichlorvos solution to ensure sample integrity. Further, the entire extraction process was carried out in ice bath maintained below 10°C. Reported procedures have employed protein precipitation (PP) [18], liquid-liquid extraction (LLE) [15,17],solid phase extraction(SPE)[7-9,12-14,16]or a combination of PP and on-line SPE[10] for sample preparation of OST and/or OSTC from different biological samples. Thus, PP was tried with trichloroacetic acid in water (5-10%) as reported earlier[18]. Although the extracts obtained were clear with adequate response for both the analytes,the recovery was inconsistent for OST at CS-1 (0.5 ng/mL) and CS-2 (1.0 ng/mL) levels. Thus,SPE was initiated on Orochem DVB-LP extraction cartridges under acidic conditions to remove plasma proteins and other interfering substances to obtain clean extracts for LC-MS/MS analysis. Use of 0.2 mL of dichlorvos solution(0.1 mg/mL in acetonitrile): water (70:30, v/v) as the eluting solvent helped in reproducible and quantitative recovery for both the analytes without drying and reconstitution steps.
3.3. Chromatography
Earlier methods have used different columns such as Nova-Pak CN HP [8], ZIC-HILIC [12], Chromatopack C18 and Synergi Hydro C18 with varying dimensions and particle size for separation of OST and OSTC from different matrices.Thus, analytical potential of four columns namely Symmetry C18 (100 mm×4.6 mm, 5 μm), ACE CN (100 mm×4.6 mm,5 μm), Cosmosil C18 (100 mm×4.6 mm, 5 μm) and Alltima C18(150 mm×4.6 mm,5 μm)were tested to achieve adequate retention and separation, short run time, symmetric peak shape and sufficient response for the analytes. Separation was tried using various combinations of acetonitrile/methanol and additives like formic acid(0.1%)and ammonium formate(5-10 mM). ACE CN column gave poor response and peak shapes, while Cosmosil C18 afforded adequate retention but the response was inadequate for both the analytes. The response obtained on Alltima C18 was satisfactory; however,the peak shape was unacceptable. Nevertheless, the best chromatographic conditions as a function of analyte peak intensity, peak shape, adequate retention and analysis run time were achieved on Symmetry C18 (100 mm×4.6 mm,5 μm) using 10 mM ammonium formate and acetonitrile(30:70, v/v) as the mobile phase under isocratic conditions(Fig.2). At the same time it afforded baseline separation(resolution factor Rs, 1.1) of the analytes within 2.0 min, with a retention time of 1.56 and 1.11 min for OST and OSTC respectively. The chromatographic run time achieved in the present work was the shortest compared to all previous assays except one report [16], which had a run time of 1.0 min.Further, the reproducibility in the measurement of retention time for both the analytes,expressed as%CV was ≤0.7%for more than 100 injections on the same column. The on-column loading of analytes for an injection volume of 10 μL at ULOQ was much less as compared to other methods (Table 1). The deuterated internal standards used in the study helped in overall assay performance and the accuracy of the data.Moreover, deuterated internal standards had similar extraction recovery as the non-labeled analytes.
3.4. Validation results
3.4.1. Assay performance and carryover
The precision (% CV) for system suitability test was in the range of 0.04-0.57% for the retention time and 0.81-1.98%for the area response of both the analytes and ISs. The signal to noise ratio for system performance was ≥30 for both the analytes and ISs. Autosampler carry-over evaluation was performed to ensure that it does not affect the accuracy and the precision of the proposed method. There was negligible carryover (≤3.3% for OST and≤4.5% for OSTC of LLOQ area) observed in extracted blank plasma after subsequent injection of highest CS at the retention time of the analytes.
3.4.2. Method selectivity
Representative MRM chromatograms of extracted blank human plasma (Fig.2A and E), blank plasma spiked with IS(Fig.2B and F),OST and OSTC at LLOQ(Fig.2C and G)and a subject sample at Cmaxafter administration of 75 mg dose of oseltamivir phosphate (Fig.2D and H) demonstrate the selectivity of the method.No endogenous compounds were found to interference at the retention time of analytes and ISs.Moreover,none of the medications commonly used by human volunteers such as acetaminophene, cetirizine, domperidone,ranitidine, diclofenac, ibuprofen, nicotine and caffeine interfered with the quantitation of analytes.

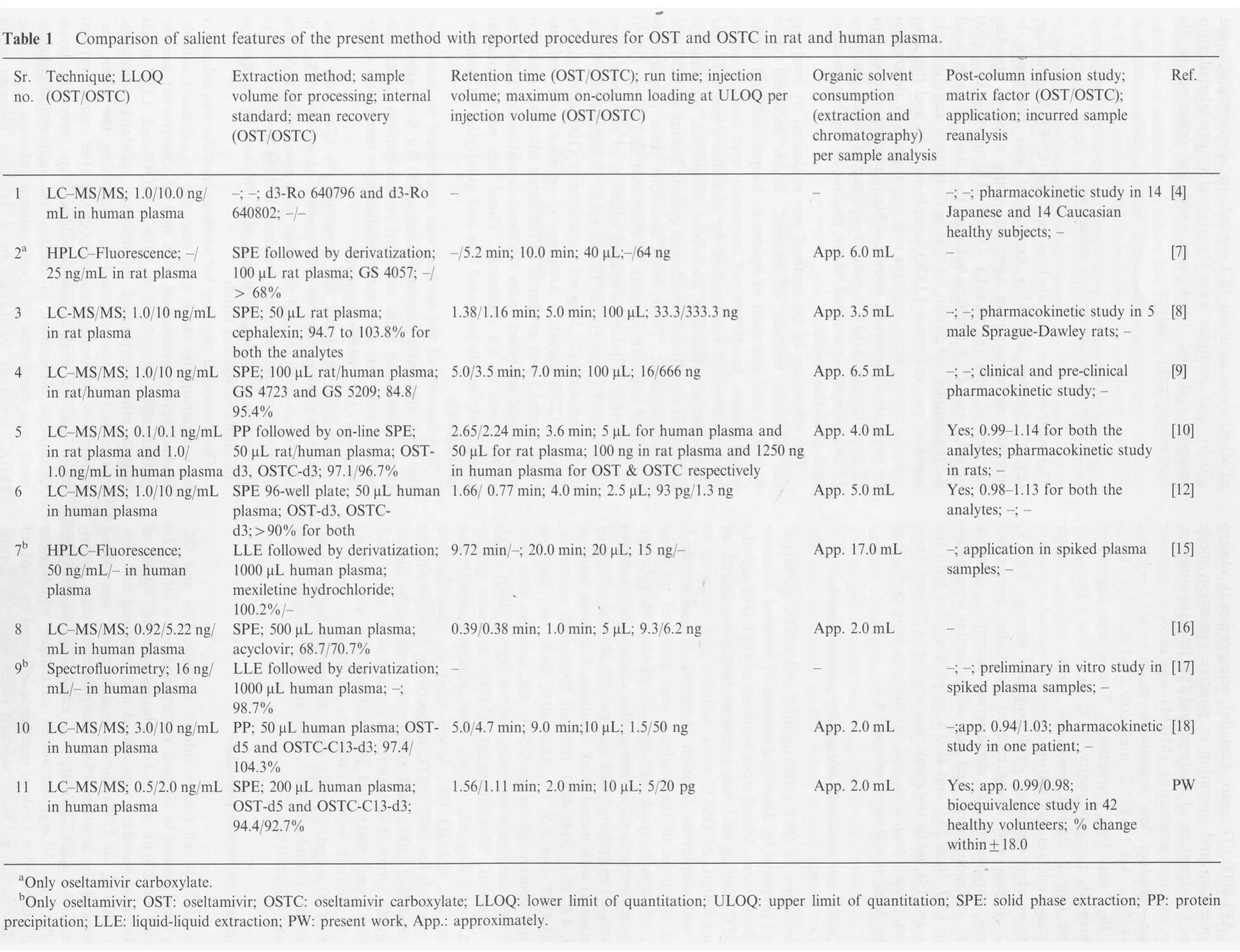
Fig.2 Representative chromatograms of (A-D) oseltamivir (m/z 313.1→166.2) and oseltamivir-d5 (IS-1, m/z 318.1→171.2) and(E-H)oseltamivir carboxylate (m/z 285.1→138.1) and oseltamivir acid-C13-d3 (IS-2, m/z 289.2→138.3) in (A) & (E) double blank plasma,(B)&(F)blank plasma spiked with IS,(C)&(G)analytes at LLOQ and IS,(D)&(H)real subject sample at Cmax after administration of 75 mg dose of oseltamivir phosphate.
3.4.3. Linearity, sensitivity, accuracy and precision
Both the analytes showed good linearities (r2≥0.9976) through the studied concentration range of 0.5-200 ng/mL for OST and 2.0-800 ng/mL for OSTC. The mean linear equations for calibration curve concentrations were y=(0.3761±0.0429)x+(0.0039±0.0060)for OST and y=(0.0549±0.0135)x+(0.0271±0.0095)for OSTC.The lowest concentration(LLOQ,0.5 ng/mL and 2.0 ng/mL)in the standard curve was measured at a signalto-noise ratio (S/N)≥30. Based on the high S/N values it was possible lower the quantitation limit by 3 folds; however, it not required based on subject sample results. The sensitivity achieved for OST and OSTC in the present work was higher compared to all other methods developed in human plasma[9,10,12,15-18]. The LOD values found at S/N≥10 (% CV less than 15) were 0.15 and 0.62 ng/mL for OST and OSTC respectively. The intra-batch and inter-batch precision and accuracy results at four QC levels are presented in Table 2.The precision(%CV)and accuracy values for intra-and inter-batch ranged from 2.42% to 5.17% and 100.2% to 103.8% for OST, and 1.87% to 4.57% and 94.5% to 102.5% for OSTC respectively.
3.4.4. Recovery and ion suppression
The extraction recovery at three QC levels is shown in Table 3.The mean extraction recovery for OST, OSTC, IS-1 and IS-2 was 94.4%, 92.4%, 93.1% and 91.9% respectively. Post column infusion chromatograms in Fig.3A-D do not show any ion suppression or enhancement at the retention time of analytes and ISs. Moreover, the internal standard normalized matrix factors varied from 0.99 to 1.02 for OST and 0.98 to0.99 for OSTC (Table 4). All the values were close to 1.0,which indicates minimum matrix interference and that the ISs efficiently compensated for any possible ion suppression or enhancement.
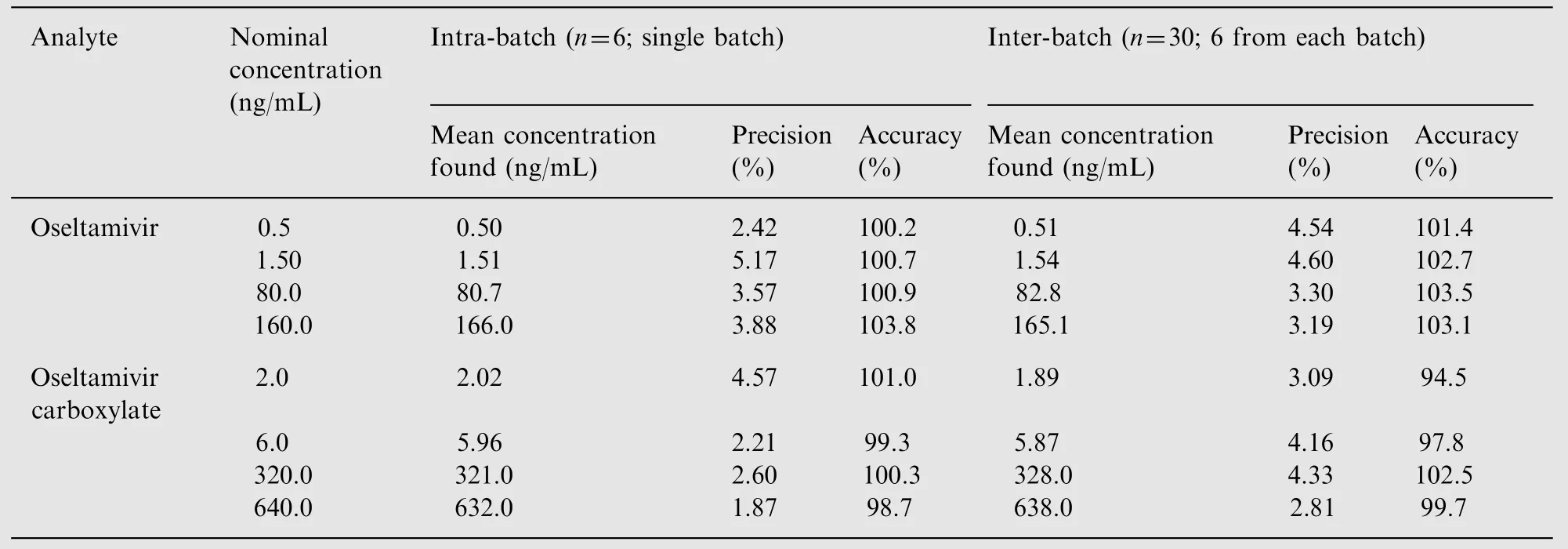
Table 2 Intra-batch and inter-batch precision and accuracy for oseltamivir and oseltamivir carboxylate.
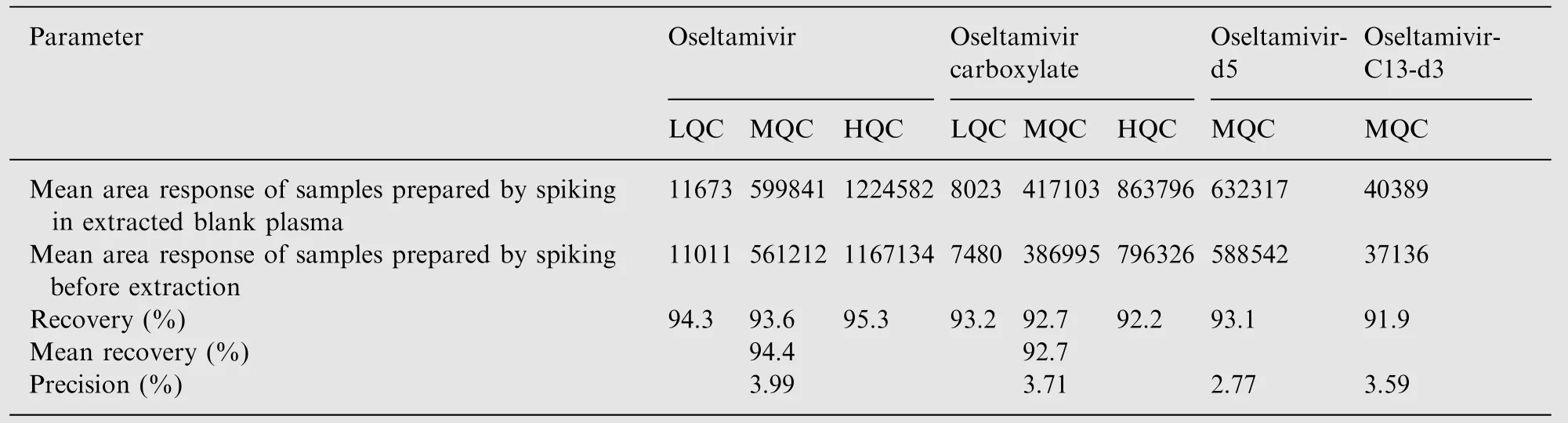
Table 3 Extraction recovery for oseltamivir and oseltamivir carboxylate from human plasma (n=6).
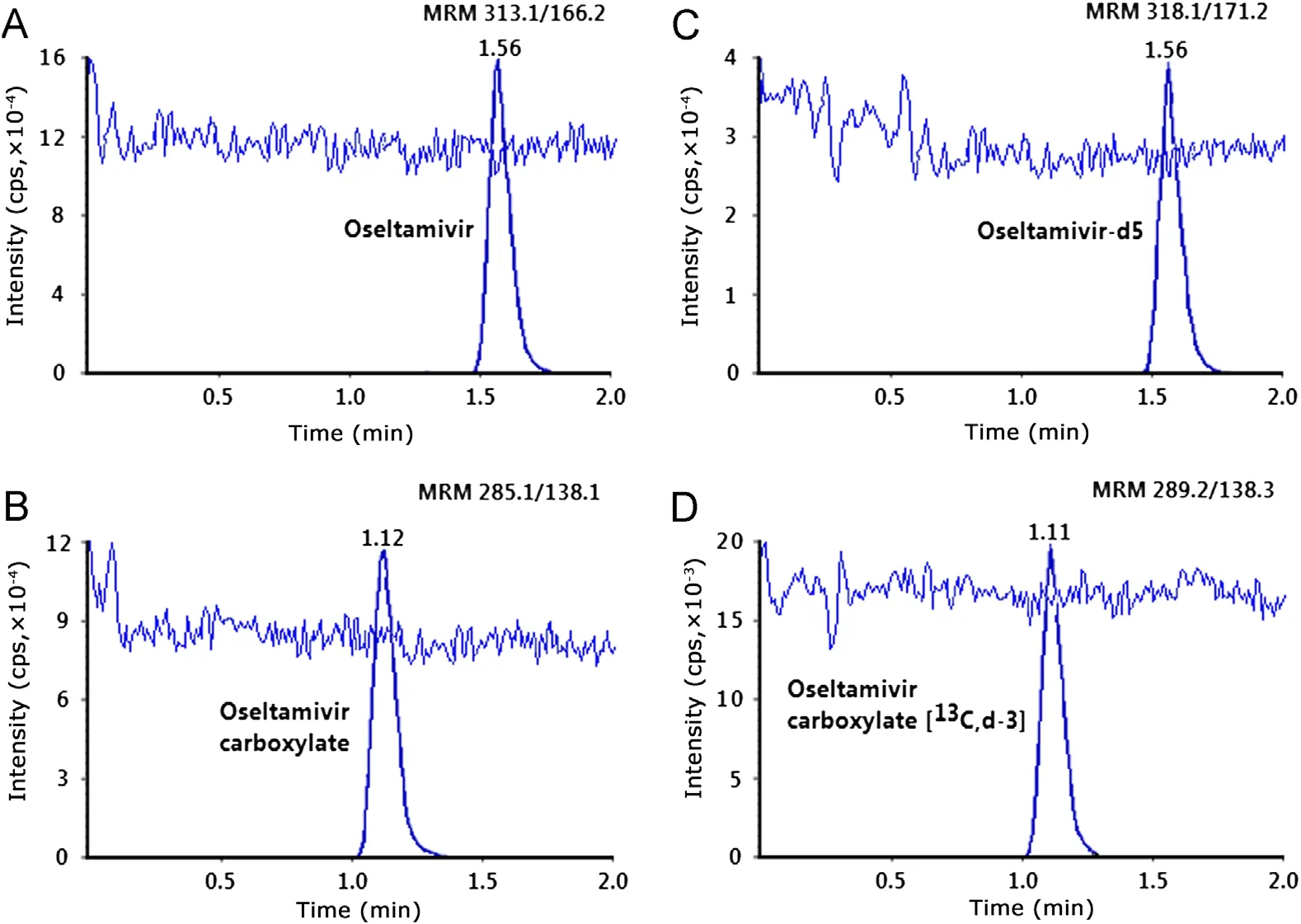
Fig.3 MRM LC-MS/MS chromatograms of blank plasma extract with post column infusion of (A) oseltamivir, (B) oseltamivir carboxylate, (C) oseltamivir-d5 and (D) oseltamivir acid-C13-d3.
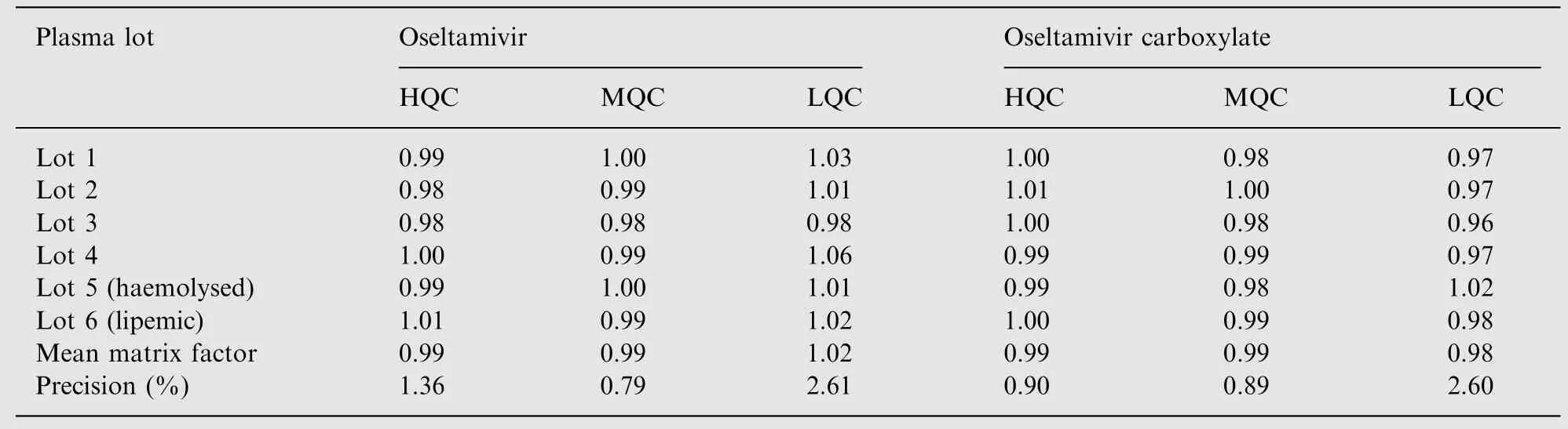
Table 4 Matrix factor (internal standard normalized) for oseltamivir and oseltamivir carboxylate.
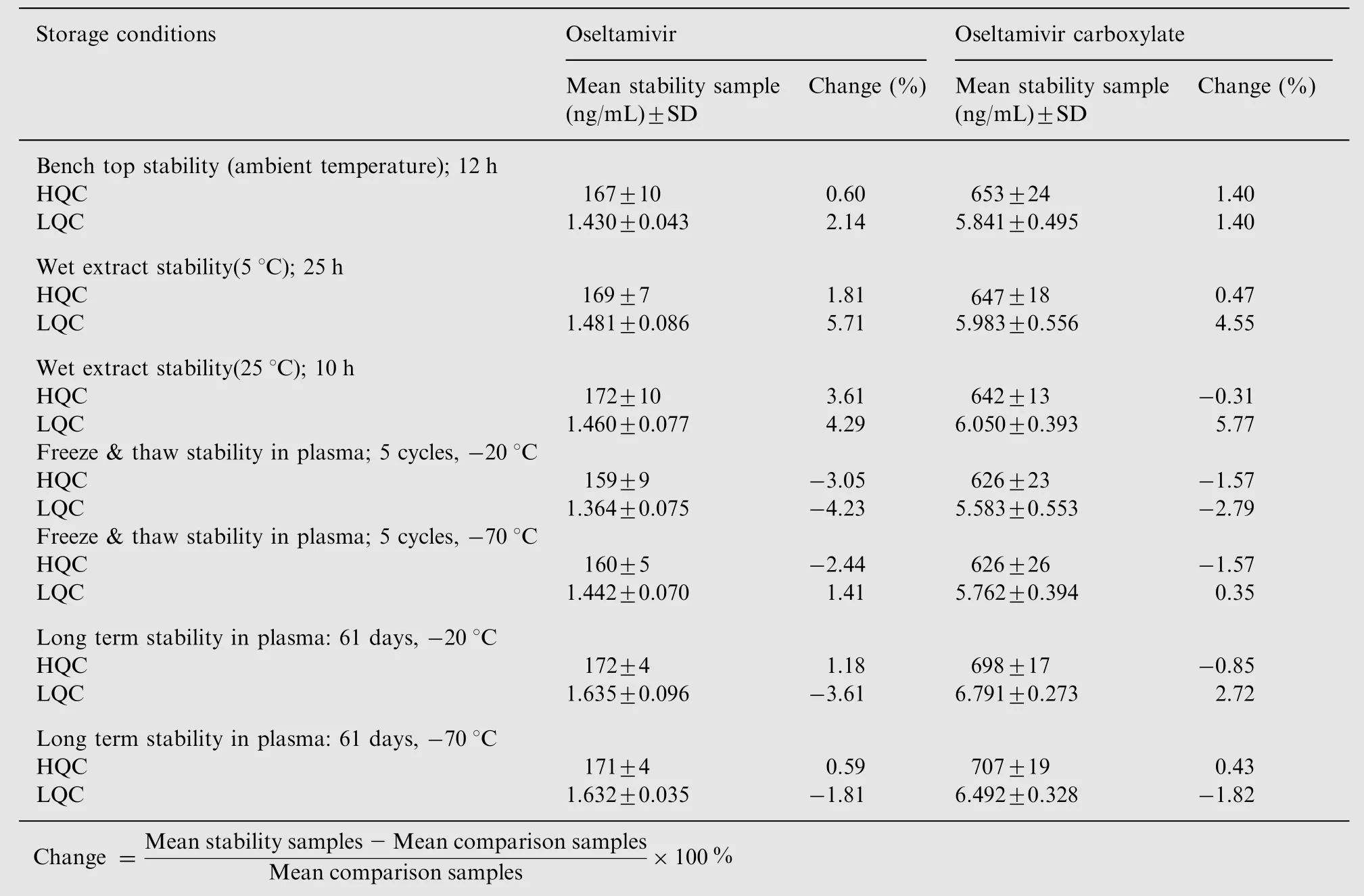
Table 5 Stability of oseltamivir and oseltamivir carboxylate under various conditions (n=6).
3.4.5. Stability results
Stability study for OST has been extensively discussed in previous reports [8,9,23]. In the present method dichlorvos was used as an esterase inhibitor to inhibit the metabolism of OST in human plasma. Samples for short-term stability remained stable up to 29 h at 25°C, while the stock solutions and working solution for long term stability were stable for a minimum of 60 days at refrigerated temperature of 5°C.The detailed results for different stability experiments in plasma are shown in Table 5.
3.4.6. Dilution reliability and method ruggedness
The precision(%CV)for dilution reliability of 1/2 and 1/10th dilution was within 1.8% to 3.5%, while the accuracy results were between 98.0% and 105.0% for both the analytes. For method ruggedness the precision (% CV) and accuracy values for two different equipments and with different analysts ranged from 0.91% to 5.35% and 95.3% to 104.8% respectively for both the analytes at three QC levels.

Fig.4 Mean plasma concentration-time profile of (A) oseltamivir and (B) oseltamivir carboxylate after oral administration of test(75 mg oseltamivir phosphate capsule of an Indian Company) and a reference (TAMIFLU®, 75 mg oseltamivir phosphate capsule from Genentech USA Inc., USA) formulation to 42 healthy Indian subjects under fed conditions.
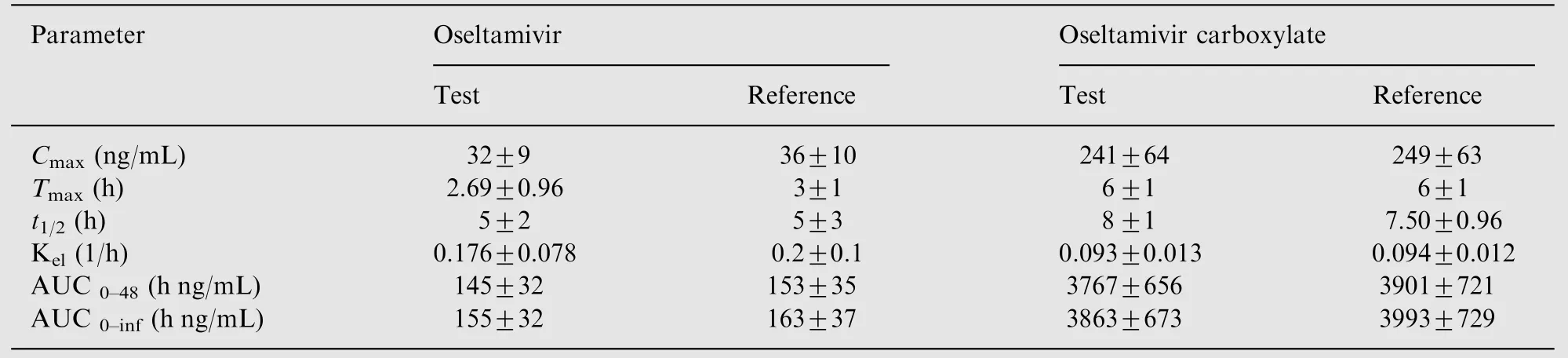
Table 6 Mean pharmacokinetic parameters of oseltamivir and oseltamivir carboxylate (Mean ±SD).
3.4.7. Bioequivalence study and incurred sample reanalysis
The validated method was successfully used to quantify OST and OSTC plasma concentration after administration of a single 75 mg oral dose of oseltamivir phosphate. Fig.4 shows the plasma concentration vs.time profile of OST and OSTC in healthy Indian subjects under fed condition. Approximately 4200 samples including the calibration, QC and subject samples were run and analyzed during a period of 12 days and the precision and accuracy were well within the acceptable limits. Table 6 summarizes the mean pharmacokinetic parameters obtained for OST and OSTC after oral administration of test and reference formulation. The mean Cmaxvalues obtained for OST in the present work were comparable with a similar study involving Japanese and Caucasian healthy volunteers [4]. However, Cmaxvalues for OSTC were significantly higher compared to Caucasian and to a lesser extent with Japanese subjects for identical dose strength. Further,Tmax,t1/2and AUC values were all on the higher side in Indian subjects compared to this study[4].This dissimilarity could be due to several factors including race of subjects, gender, type of food and others. However, the ratios of mean logtransformed parameters and their 90% confidence intervals(90.07-96.74%)were within the acceptance range of 80-125%.The precision(%CV)values for intra-subject variation ranged from 6.43% to 6.55% for Cmax, AUC0-tand AUC0-inffor both the analytes.The assay reproducibility was established by reanalysis of 151 incurred samples. Out of these, 114 samples showed % change of ±10%, while the remaining 37 samples were within±18.0%of the initial results for both the analytes.This authenticates the reproducibility of the validated assay.
4. Conclusion
A sensitive, selective and rapid method for the simultaneous determination of OST and OSTC in human plasma has been developed and fully validated as per USFDA guidelines. The efficiency of SPE and a chromatographic run time of 2.0 min per sample make it highly useful for high-throughput bioanalysis of OST and OSTC. Moreover, the present method does not involve drying and reconstitution steps during sample processing compared to other SPE procedures [8,9,16] or a derivatization step post extraction [7].The proposed method is more sensitive for both the analytes compared to all other methods developed in human plasma. The chromatographic run time is shorter compared to all other methods except one report [16]. The linear dynamic range ensures application of the method for even higher dose strength with acceptable precision and accuracy. With dilution reliability up to 2-folds,it is possible to extend the upper limit of quantification to 400 and 1600 ng/mL for OST and OSTC respectively. The validated method has shown acceptable precision and accuracy for their simultaneous quantification in human plasma in a clinical study. Incurred sample reanalysis with 151 samples demonstrates the reproducibility in the measurement of subject samples.
The authors are thankful to the Chief Operating Officer,Mr. E. Venu Madhav and directors, Mr. Apurva Shah and Mr. Binoy Gardi of Veeda Clinical Research Pvt. Ltd. (India)for providing infrastructure facility to carrying out this work.
[1] A. Moscona, Neuraminidase inhibitors for influenza, New Engl.J. Med. 353 (2005) 1363-1373.
[2] G.Z.He,J.W.Massarella,P.Ward,Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802,Clin. Pharmacokinet. 37 (1999) 471-484.
[3] R. Dutkowski, J.R. Smith, B.E. Davies, Safety and pharmacokinetics of oseltamivir at standard and high doses, Int. J. Antimicrob. Agents 35 (2010) 461-467.
[4] J.J. Schentag, G. Hill, T. Chu, et al., Similarity in pharmacokinetics of oseltamivir and oseltamivir carboxylate in Japanese and Caucasian subjects, J. Clin. Pharmacol. 47 (2007) 689-696.
[5] J.W. Massarella, G.Z. He, A. Dorr, et al., The pharmacokinetics and tolerability of the oral neuraminidase inhibitor oseltamivir(Ro 64-0796/GS4104) in healthy adult and elderly volunteers,J. Clin. Pharmacol. 40 (2000) 836-843.
[6] W.B. Dreitlein, J. Maratos, J. Brocavich, Zanamivir and oseltamivir: two new options for the treatment and prevention of influenza, Clin. Ther. 23 (2001) 327-355.
[7] E.J. Eisenberg, K.C. Cundy, High-performnace liquid chromatographic determination of GS4071, a potent inhibitor of influenza neuraminidase, in plasma by precolumn fluorescence derivatization with naphthalenedialdehyde, J. Chromatogr B 716 (1998)267-273.
[8] Q. Chang, M.S.S. Chow, Z. Zuo, Studies on the influence of esterase inhibitor to the pharmacokinetic profiles of oseltamivir and oseltamivir carboxylate in rats using an improved LC/MS/MS method, Biomed Chromatogr. 23 (2009) 852-857.
[9] H. Wiltshire, B. Wiltshire, A. Citron, et al., Development of a high-performance liquid chromatographic-mass spectrometric assay for the specific and sensitive quantification of Ro 64-0802,an anti-influenza drug, and its pro-drug, oseltamivir, in human and animal plasma and urine, J. Chromatogr. B 745 (2000)373-388.
[10] K. Heinig, F. Bucheli, Sensitive determination of oseltamivir and oseltamivir carboxylate in plasma, urine, cerebrospinal fluid and brain by liquid chromatography-tandem mass spectrometry,J. Chromatogr. B 876 (2008) 129-136.
[11] K. Heinig, T. Wirz, F. Bucheli, et al., Determination of oseltamivir (Tamiflu®) and oseltamivir carboxylate in dried blood spots using offline or online extraction, Bioanalysis 3 (2011)421-437.
[12] N. Lindeg ˚ardh, W. Hanpithakpong, Y. Wattanagoon, et al.,Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of oseltamivir and its metabolite oseltamivir carboxylate in plasma,saliva and urine,J. Chromatogr. B 859 (2007) 74-83.
[13] C. Fuke, Y. Ihama, T. Miyazaki, Analysis of oseltamivir active metabolite, oseltamivir carboxylate, in biological materials by HPLC-UV in a case of death following ingestion of Tamiflu®,Legal Med. 10 (2008) 83-87.
[14] G. Bahrami, B. Mohammadia, A. Kiani, Determination of oseltamivir carboxylic acid in human serum by solid phase extraction and high performance liquid chromatography with UV detection, J. Chromatogr. B 864 (2008) 38-42.
[15] Z. Aydogmus, S. Caglar, S. Toker, RP-HPLC method for determination of oseltamivir phosphate in capsules and spiked plasma, Anal. Lett. 43 (2010) 2200-2209.
[16] R. Kanneti, D. Bhavesh, D. Parmar, et al., Development and validation of a high-throughput and robust LC-MS/MS with electrospray ionization method for simultaneous quantitation of oseltamivir phosphate and its oseltamivir carboxylate metabolite in human plasma for pharmacokinetic studies, Biomed. Chromatogr. 25 (2011) 727-733.
[17] M.I. Walash, F. Belal, N. El-Enamy, et al., Spectrofluorimetric determination of oseltamivir phosphate through derivatization with o-phthalaldehyde application to pharmaceutical preparations with a preliminary study on spiked plasma samples,Luminescence. 27 (6) (2012) 511-518.
[18] W. Kromdijk, H. Rosing, M.P.H. van den Broek, et al., Quantitative determination of oseltamivir and oseltamivir carboxylate in human fluoride EDTA plasma including the ex vivo stability using high-performance liquid chromatography coupled with electropsray ionization tandem mass spectrometry, J. Chromatogr. B 892 (2012) 57-63.
[19] Guidance for Industry, Bioanalytical Method Validation,US Department of Health and Human Services, Food and Drug Administration Centre for Drug Evaluation and Research (CDER), Centre for Veterinary Medicine (CVM),〈http://www.fda.gov/downloads/〉 Drugs/GuidanceCompliance RegulatoryInformation/Guidances/ucm070107.pdf, 2001 (assessed 12.07.12).
[20] V.B. Ravi, J.K. Inamadugu, N.R. Pilli, et al., Simultaneous determination of telmisartan and amlodipine in human plasma by LC-MS/MS and its application in a human pharmacokinetic study, J. Pharm. Anal. 2 (2012) 319-326.
[21] C.R. Mallet, Z. Lu, J.R. Mazzeo, A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts, Rapid. Commun. Mass Spectrom. 18 (2004)49-58.
[22] Guidance for Industry: ICH E6 Good Clinical Practice, US Department of Health and Human Services, Food and Drug Administration, Centre for Drug Evaluation and Research(CDER),Centre for Biologics Evaluation and Research(CBER),〈http://www.fda.gov/ downloads/regulatoryinformation/guidances/ucm129515.pdf〉, 1996 (assessed 12.07.12).
[23] M.Yadav,P.S.Shrivastav,Incurred sample reanalysis:a decisive tool in bioanalytical research, Bioanalysis 3 (2011) 1007-1024.
[24] N.Lindegardh,G.R.Davies,T.T.Hien,et al.,Rapid degradation of oseltamivir phosphate in clinical samples by plasma esterases,Antimicrob. Agents Chemother. 50 (2006) 3197-3199.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Cover Figure
- Improved reversed phase liquid chromatographic method with pulsed electrochemical detection for tobramycin in bulk and pharmaceutical formulation
- Fluorescence spectroscopy of osthole binding to human serum albumin
- Release test of alliin/alliinase double-layer tablet by HPLC—Allicin determination
- Accurate quantitation standards of glutathione via traceable sulfur measurement by inductively coupled plasma optical emission spectrometry and ion chromatography
- Determination and stress studies on YK-1101,a potential histone deacetylase, by HPLC-UV and HPLC-TOF/MS methods