典型单环和双环芳烃加氢热力学分析
2013-12-23侯朝鹏李永丹夏国富李明丰
侯朝鹏,李永丹,夏国富,李明丰
(1. 中国石化 石油化工科学研究院,北京 100083;2. 天津大学 化工学院,天津 300072)
柴油中芳烃含量高不仅会降低油品的质量和十六烷值,还会增加柴油燃烧废气中的颗粒排放物,因而,芳烃加氢受到广泛关注[1-3]。油品中的芳烃按芳环的数量主要分为4类:单环芳烃、双环芳烃、三环芳烃和多环芳烃。在实验研究中,通常选取苯及其同系物作为单环芳烃的模型反应物,萘及其同系物作为双环芳烃的模型反应物。文献[4-5]曾报道了几种芳烃及其加氢产物的热力学数据。Poling等[5-7]为解决芳烃热力学数据缺乏的问题,将基团贡献法用于估算某些芳烃及其加氢产物的标堆焓、熵和理想气体热容。Frye等[8-9]采用气相体系测定了不同温度和压力下几种芳烃/氢气混合物的平衡组成,所选用的芳烃有联苯、茚、萘、菲、二氢苊和芴。Jaffe[10]利用文献[4]中的数据总结了烃类加氢放热的规律,他认为不同种类烃的C—C键断裂所释放的能量不同,文献[11]所报道的对苯加氢热力学的计算结果与其一致。对含有支链的苯系芳烃,平衡常数随侧链数和每一个侧链上碳原子数的增加而减小;萘系芳烃与苯系芳烃类似[12-14]。一般来说,含一个以上环的芳烃,其加氢是一个一个芳环逐步进行的。在传统加氢条件下,第一个芳环的加氢平衡常数一般较高[12,15-16]。上述文献均没有考虑实际的反应过程和复杂的实验条件。
对于实际的生产过程,可选择几种典型芳烃的加氢热力学加以考虑,以寻找热力学上合适的反应条件和操作区域,再综合选择它们的合适操作区域的公共部分,使不同的芳烃处于热力学可行的加氢条件范围之内。在实验研究中,一般会针对反应过程选择不同的模型反应物,而各种典型芳烃的加氢热力学平衡计算会为芳烃加氢反应直接提供相关的热力学允许的操作条件。
本工作利用热力学数据库软件HSC-Chemistry 4.0对几种典型的芳烃加氢反应热力学平衡进行讨论,充分考虑了反应条件的变化对芳烃加氢转化率的影响,直接提供了热力学平衡允许的操作条件。
1 热力学计算依据及数据基础
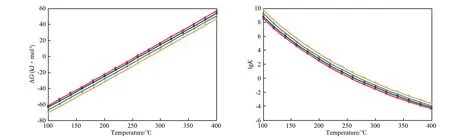
图1 几种单环芳烃加氢饱和反应的ΔG和lgK随温度的变化Fig.1 ΔG and lgK vs temperature in the saturate hydrogenation of several monocyclic aromatics.
利用商用软件HSC-Chemistry4.0(Outokumpu公司产品)进行热力学计算,各种物质的热力学数据由软件的数据库引出。所有气体均被认为是理想气体;不特别指明时,所有反应以每mol主反应物为基准,反应热的单位为kJ/mol;假定所有反应体系均为封闭体系,不特别指明时,压力指绝对压力,计算所得结果均为达到化学平衡时的组成,转化率为平衡转化率。苯加氢生成环己烷较为简单,因此本工作采用苯的平衡转化率对该过程进行描述;而萘的加氢产物较为复杂,为了表达方便,采用含氢基的平衡组成对该过程进行描述。
2 计算结果
2.1 单环芳烃加氢反应
一般常用的单环芳烃模型反应物有苯、甲苯、乙苯和二甲苯,以及它们的同分异构体和同系物。几种单环芳烃加氢饱和反应的反应自由能变(ΔG)和lgK(K为反应平衡常数)随温度的变化见图1。从图1可见,这些反应的共同特点是:在100~400 ℃内,ΔG随反应温度的升高近似线性增大,K随反应温度的升高而降低。反应在低于250~300 ℃时为自发过程,反应温度越低,ΔG越小,反应越倾向于自发进行。从图1还可见,在相同反应温度下,4种芳烃加氢反应的ΔG随烷基侧链上碳原子数的增加而增大,K随烷基侧链上碳原子数的增加而减小。这与文献[12-14]报道的结果一致。
由于这些单环芳烃的热力学反应过程及数据的规律性较为相似,因此,以苯为例进行计算,其他单环芳烃的计算结果与其类似。苯不仅是一种重要的化工原料,且在研究中经常作为单环芳烃加氢过程的模型反应物[17-27]。气相苯的完全加氢产物是环己烷,该反应是体积缩小反应,增加系统压力有利于苯加氢转化率的提高;且该反应为强放热反应,苯平衡转化率随温度的升高而降低。影响苯平衡转化率的因素较为复杂,本工作主要计算温度、压力和氢气与苯的摩尔比(氢烃比)对苯平衡转化率的影响。
温度和氢烃比对苯加氢生成环己烷的平衡转化率的影响见图2。由图2可看出,在各氢烃比下,平衡转化率随温度的升高而降低。在氢烃比小于3时,由于每一个苯分子加氢生成环己烷要消耗3个分子的氢气,如果氢气量不足,会使苯转化不完全;当氢烃比大于3时,氢气过量,加氢后还有过量的氢气存在,此时苯转化率较高,且随氢烃比的增加,平衡转化率大于99.0%和99.9%的温度范围(等高线内侧的两条线内的区域,等高线是响应面在x-y平面上的投影)越来越大。

图2 温度和氢烃比对苯加氢生成环己烷的平衡转化率的影响Fig.2 Effects of temperature and n(H2)∶n(aromatic) on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
在实际反应过程中,苯加氢反应一般在较高的氢烃比下操作。温度和压力对苯加氢生成环己烷的平衡转化率的影响见图3。

图3 温度和压力对苯加氢生成环己烷的平衡转化率的影响Fig.3 Effects of temperature and pressure on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
由图3可看出,压力为0.1 MPa时,在200 ℃以下,苯的平衡转化率很高,在99.9%以上。随温度的升高,由于热力学平衡的影响,苯的平衡转化率逐渐降低;温度高于350 ℃时,苯的平衡转化率已很低。在每个温度点,苯的平衡转化率随压力的增加而增大,且随压力的增加,平衡转化率超过99.9%和99.0%的温度范围越来越大,如在5.0 MPa时,苯的平衡转化率超过99.9%的温度范围比0.1 MPa时升高了约100 ℃。
图4是常规工业操作压力(5.0 MPa)下,温度和氢烃比对苯加氢生成环己烷的平衡转化率的影响。由图4可看出,在氢烃比低于3时,由于氢气不足,平衡转化率较低;且随反应温度的升高,平衡转化率明显降低,这与图1中ΔG随温度的升高而增加,K随温度的升高而降低的趋势一致。
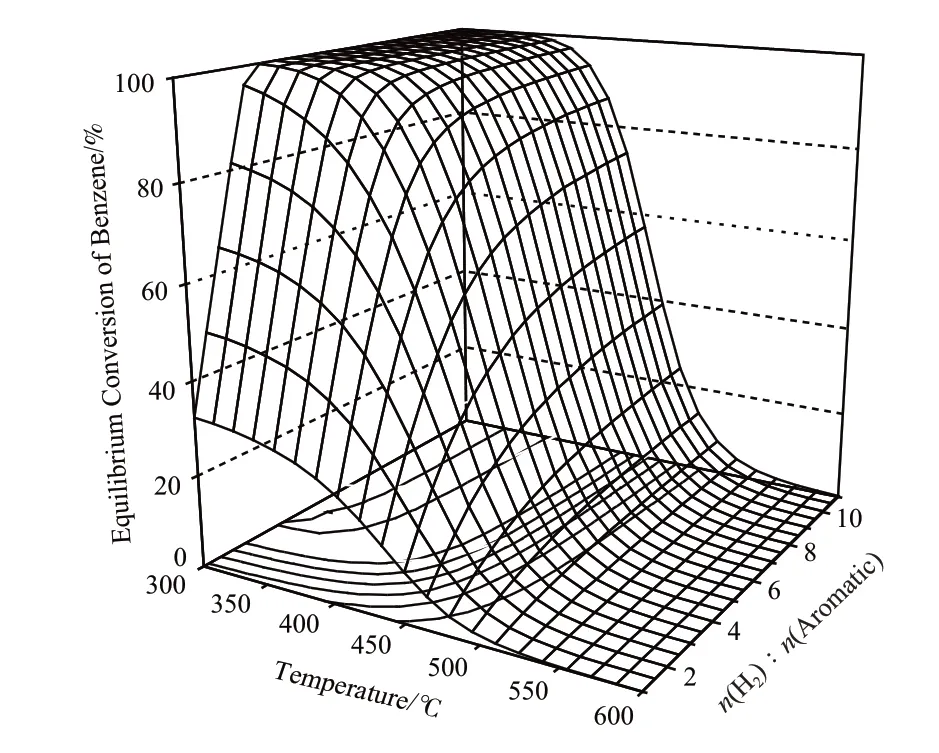
图4 温度和氢烃比对苯加氢生成环己烷的平衡转化率的影响Fig.4 Effects of temperature and n(H2)∶n(aromatic) on the equilibrium conversion of benzene in its hydrogenation to cyclohexane.
从以上计算结果可知,温度、氢烃比和压力是影响苯平衡转化率的重要因素。在选定反应温度时,提高反应系统的压力和氢烃比,都可使苯加氢生成环己烷的平衡转化率增大。
在以苯为模型反应物的芳烃加氢系统中,由于使用的催化剂不同,反应的活性温度范围差异很大。不同压力下Ni催化剂催化苯加氢反应的转化率与温度的关系见图5。由图5可见,若无Ni催化剂,苯加氢转化率很低;采用Ni催化剂后,苯加氢转化率明显增加,且增加压力对提高苯加氢转化率有利[24]。
因此,为了寻找苯加氢合适的操作条件,必须充分综合考虑催化剂、温度、压力和氢烃比等因素。图2~4提供了苯加氢反应操作条件范围的具体数值,这些数据为文献[16]提供了有益的补充。由于图1中的几种主要单环芳烃模型物加氢饱和反应的ΔG和lgK随温度的变化情况一致,由此可以推测这几种单环芳烃加氢反应的实验数据也会与苯加氢反应的实验数据呈相似的规律。
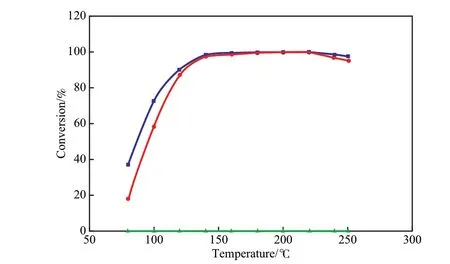
图5 不同压力下Ni催化剂催化苯加氢反应的转化率与温度的关系Fig.5 Conversion of benzene vs temperature under different pressure on Ni catalyst.
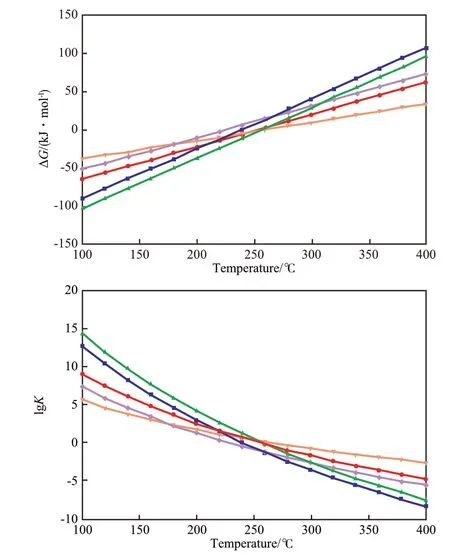
图6 ΔG和lgK随温度的变化曲线Fig.6 ΔG and lgK vs temperature.
2.2 双环芳烃加氢反应
双环芳烃模型反应物的代表主要为萘。气相萘加氢过程较复杂,一般有两种稳定的加氢产物四氢萘和十氢萘,其中十氢萘有两种同分异构体:顺式十氢萘和反式十氢萘。主要反应的方程式如下:
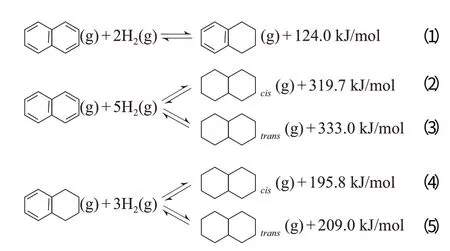
许多研究者[28-40]选择萘作为芳烃加氢过程的模型反应物,也有研究者[41-50]选择四氢萘。不同反应的ΔG和lgK随温度的变化见图6。从图6可见,这些反应的共同特点是:在100~400 ℃内,ΔG随反应温度的升高近似线性单调升高,K随反应温度的升高单调降低;在约250 ℃以下,ΔG为负值,说明这些反应在低于250 ℃时为自发过程,且反应温度越低,ΔG越小,反应越倾向于自发进行。从图6还可见,不同反应的ΔG随温度变化的趋势有差别,其中萘加氢生成四氢萘的ΔG随温度的变化幅度最小。
萘体系加氢为体积缩小的反应,增加系统压力有利于萘加氢转化率的提高;提高氢气分压也有利于萘的转化,且反应为强放热反应,萘体系的平衡转化率随温度的升高而降低。考虑到通常的反应状况,选择在氢气过量下计算萘加氢过程的热力学平衡组成。温度和压力对萘体系加氢产物分布的影响见图7。由图7可见,对应于每个系统压力,反应过程中的组分都很复杂,一般是氢气、萘、四氢萘和十氢萘的混合物。从图7a可见,在该体系中,低温下萘可以完全加氢转化,但在不同压力下,萘完全转化的温度不同,随压力的增加,萘完全加氢转化的温度范围越来越宽。在高温下,对应于每个压力,萘的平衡转化率随温度的增加而降低。而随压力的升高,对应较低萘平衡组成的温度区逐渐变宽,说明系统压力的升高有利于萘加氢,如在0.1 MPa下,萘接近完全转化的温度约为200 ℃;而在5.0 MPa时,萘接近完全转化的温度升至400 ℃。
从图7b可看出,在该体系中,对应每个压力,随温度的升高,四氢萘的平衡组成存在一个峰值,在压力为0.1 MPa时,对应温度约为250 ℃。随系统压力的升高,对应四氢萘平衡组成峰值的温度逐渐向高温偏移。在图7b中,由于计算步长的设计问题,导致数据的连续性不佳,但图内数据仍反映了四氢萘的平衡组成随反应条件变化的趋势。
从图7c可看出,在该体系中,对应每个压力,随温度的升高,反式十氢萘的平衡组成不断降低。而随压力的升高,有利于生成反式十氢萘的温度区域逐渐变宽,说明系统压力的升高有利于反式十氢萘的生成。
从图7d可看出,在该体系中,对应每个压力,随温度的升高,顺式十氢萘的平衡组成存在一个峰值,在压力为0.1 MPa时,峰值对应温度约为200 ℃;在压力为5.0 MPa时,峰值对应温度约为300 ℃。随系统压力的升高,峰值对应温度逐渐向高温偏移,且峰值逐渐增大。通过比较图7c和图7d可看出,在热力学上萘加氢生成反式十氢萘比生成顺式十氢萘有利,如在0.1 MPa下,反式十氢萘在含氢基的体系中的平衡组成最高可达约16.3%(φ),而顺式十氢萘的平衡组成最高只有0.78%(φ)。

图7 温度和压力对萘体系加氢产物分布的影响Fig.7 Effects of temperature and pressure on the equilibrium composition of the products in naphthalene hydrogenation.
在以气相萘为模型反应物的芳烃加氢系统中,由于使用的催化剂不同,反应过程的活性温度范围可能差异很大。对于一定条件的进料,为了更好地获得某种产物,可协调反应温度和压力以得到合适的操作条件。
3 计算结果与讨论
本工作利用三维视图的形式对多因素进行了描绘和考察,结果表达明了简洁。虽然没有进行各种芳烃及其所有加氢产物的热力学计算,但所选几种芳烃都是生产和实验研究中经常使用的典型模型反应物。以上计算结果有助于研究者寻找典型芳烃加氢在热力学上合适的操作区域,作为实验中选择反应条件的依据。对于实际操作过程,可以选择它们的合适操作区域的公共部分,在热力学上保证不同芳烃加氢过程的可行性。特别是实验操作过程中,一般会针对反应过程选择具体的模型反应物,而这些典型芳烃的热力学计算会直接提供相关热力学上所允许的反应温度、氢烃比和系统压力等操作条件,从而使整个研究过程得以简化。
芳烃加氢过程的强放热效应是由加氢过程中氢原子的加入破坏了芳烃有机物的共轭双键引起的。根据化学平衡原理,随温度的升高,K减小,反应平衡体系中芳烃平衡组成增加,芳烃加氢产物的平衡组成减小[16]。温度是影响芳烃加氢反应热力学平衡的重要因素。从热力学计算可知,芳烃加氢是可逆反应,从图2~4和图6可看出,温度对芳烃加氢反应有重要影响;并且由于芳烃加氢反应是强放热反应,平衡转化率随温度的升高而降低。虽然从热力学平衡角度低温有利于芳烃的加氢转化,但由于动力学的限制,实际芳烃加氢转化率也会受到很大限制。在高温下,芳烃加氢反应在动力学上通常会进行得非常快,但在热力学上会受到热力学平衡的限制,芳烃加氢的转化率反而会降低[51]。因此,必须针对催化剂和工艺选取合适的操作温度。在实际实验操作和工业生产中,要充分考虑工况中的问题,如反应过程中热点的出现会使反应床层温度升高,从平衡角度看,必然对提高芳烃加氢的转化率不利。因此,如果在反应过程中提高反应压力和氢烃比,则可使保持高芳烃转化率的可操作温度范围变宽,从而保证生产的顺利进行。在生产过程中,催化剂会部分失活,有时需要提高反应温度使生产进行下去,要生产低芳烃含量的油品和溶剂,则必须提高反应系统的压力和氢烃比。
4 结论
1)利用HSC-Chemistry4.0软件对几种常见芳烃加氢反应进行了热力学计算,考虑了温度、压力和氢烃比3个因素对芳烃加氢过程中芳烃转化率的影响。根据化学平衡原理,随温度的升高,热力学平衡常数K减小,反应平衡体系中芳烃平衡组成增大,芳烃加氢产物的平衡组成减小。
2)对于苯加氢体系,从温度、氢烃比和压力3个因素对芳烃加氢转化率的三维响应面及其等高线图可看出,等高线99.9%甚至99.0%以内的区域所对应的温度、氢烃比和压力为芳烃加氢高转化率的较佳操作条件。
3)在萘加氢体系中,产物较为复杂,对应不同温度和压力,它们的含氢基平衡组成不同,各种产物的选择性也不同。这些计算为选择该过程操作参数和条件提供了参考和基础数据。
[1] 李大东,蒋福康. 清洁燃料生产技术的新进展[J]. 中国工程科学,2003,5(3):6 - 14.
[2] 李大东. 我国环境友好汽车燃料的发展方向[J]. 中国石化,2002(4):22 - 25.
[3] 闵恩泽,李大东. 环境友好石油炼制技术的进展[J]. 石油化工动态,1997,5(3):6 - 11.
[4] Stull D R,Westrum E F,Sinke G C. Chemical Thermodynamics of Organic Compounds[M]. New York:Wiley,1969:235 - 400.
[5] Poling B E,Prausnitz J M,O’Connell J P. The Properties of Gases and Liquids[M]. New York:McGraw-Hill,2001:6.1 - 6.34.
[6] Shaw R,Golden D M,Benson S W. Thermochemistry of Some Six-Membered Cyclic and Polycyclic Compounds Debated to Coal[J]. J Phys Chem,1977,81(12):1716 - 1729.
[7] Stein S,Golden D M,Benson S W. Prediction Scheme for Thermodynamical Properties of Polycyclic Aromatic Hydrogenation[J]. J Phys Chem,1977,81(2):314 - 316.
[8] Frye C G. Equilibria in the Hydrogenation of Polycyclic Aromatics[J]. J Chem Eng Data,1962,7(4):592 - 595.
[9] Frye C G,Weitkamp A W. Equilibrium Hydrogenations of Multi-Ring Aromatics[J]. J Chem Eng Data,1969,14(3):372 - 376.
[10] Jaffe S B. Kinetics of Heat Release in Petroleum Hydrogenation[J]. Ind Eng Chem Process Des Dev,1974,13(1):34 - 39.
[11] 李宣文,黄志渊,译. 接触催化:工业催化剂原理、制备及其应用[M]. 北京:石油工业出版社,1984:7.
[12] Le Page J F. Applied Heterogeneous Catalysis:Design,Manufacture,Use of Solid Catalysts[M]. Paris:Technip,1987:78 - 80.
[13] Wilson M F,Fisher I P,Kriz J F. Hydrogenation and Extraction of Aromatics from Oil Sands Distillates and Effects on Cetane Improvement[J]. Energy Fuels,1987,1(6):540 - 544.
[14] Gully A J,Balard W P. Hydrogenation of Catalytic Cracking Charge Stocks[M]//McKetta J J,ed. Advances in Petroleum Chemistry and Refining. New York:Interscience Publishers,1963,7:241 - 282.
[15] Stanislaus A,Copper B H. Aromatic Hydrogenation Catalysis:A Review[J]. Catal Rev Sci Eng,1994,36(1):75 - 123.
[16] 李大东. 加氢处理工艺与工程[M]. 北京:中国石化出版社,2004:119 - 129.
[17] Tsai K Y,Wang I,Tsai T C. Zeolite Supported Platinum Catalysts for Benzene Hydrogenation and Naphthalene Isomerization[J]. Catal Today,2011,166(1):73 - 78.
[18] Boudjahem A G,Bouderbala W,Bettahar M. Benzene Hydrogenation over Ni-Cu/SiO2Catalysts Prepared by Aqueous Hydrazine Reduction[J]. Fuel Process Technol,2011,92(3):500 - 506.
[19] Abu Bakar N H H,Bettahar M M,Abu Bakar M,et al. Low Temperature Activation of Pt/Ni Supported MCM-41 Catalysts for Hydrogenation of Benzene[J]. J Mol Catal A:Chem,2010,333(1/2):11 - 19.
[20] Savva P G,Goundani K,Vakros J,et al. Benzene Hydrogenation over Ni/Al2O3Catalysts Prepared by Conventional and Sol-Gel Techniques[J]. Appl Catal,B,2008,79(3):199 - 207.
[21] Reshetnikov S I, Ivanov E A, Startsev A N. Benzene Hydrogenation in the Thiophene Presence over the Sulfide Ni-Mo/γ-Al2O3Catalyst Under Periodic Operation:Kinetics and Process Modeling[J]. Chem Eng J,2007,134(1/3):100 - 105.
[22] 孙书田. NCG-98H新型苯加氢催化剂的工业应用[J]. 化学工业与工程技术,2010,31(2):47 - 49.
[23] 宋华,唐龙,武显春. 负载型Ni-B/γ-Al2O3非晶态合金催化剂的结构及苯加氢性能[J]. 现代化工,2010,30(1):54 - 56.
[24] 侯朝鹏. FC制备镍金属催化剂苯加氢反应和抗噻吩能力的研究[D]. 天津:天津大学,2003.
[25] 赵会吉,张津林,白锐,等. 新型固定床Raney Ni催化剂的苯加氢活性及耐硫性[J]. 石油化工,2007,36(7):653 -658.
[26] 傅送保,段燕凌,任军,等. 镍基均相络合催化剂苯加氢反应动力学研究[J]. 石油化工,2006,35(8):745 - 748.
[27] 周志明,李卓,程振民,等. 在Pd/-Al2O3催化剂上气相苯加氢反应动力学研究[J]. 石油化工,2003,32(5):392 - 397.
[28] Ito K,Kogasaka Y,Kurokawa H,et al. Preliminary Study on Mechanism of Naphthalene Hydrogenation to Form Decalins via Tetralin over Pt/TiO2[J]. Fuel Process Technol,2002,79(1):77 - 80.
[29] Ren Shibiao,Zhang Ping,Shui Hengfu,et al. Promotion of Ni/SBA-15 Catalyst for Hydrogenation of Naphthalene by Pretreatment with Ammonia/Water Vapour[J]. Catal Commun,2010,12(2):132 - 136.
[30] Du Mingxian,Qin Zhangfeng,Ge Hui,et al. Enhancement of Pd-Pt/Al2O3Catalyst Performance in Naphthalene Hydrogenation by Mixing Different Molecular Sieves in the Support[J].Fuel Process Technol,2010,91(11):1655 - 1661.
[31] Li Feng,Yi Xiaodong,Zheng Jinbao,et al. A Pretreatment Method of Ni/γ-Al2O3Catalyst for Naphthalene Hydrogenation[J]. Catal Commun,2009,11(4):266 - 271.
[32] Chen Honglin,Yang Hong,Omotoso O,et al. Ring,Contribution of Hydrogen Spillover to the Hydrogenation of Naphthalene over Diluted Pt/RHO Catalysts[J]. Appl Catal,A,2009,358(2):103 - 109.
[33] Sebastián D,Bordejé E G,Calvillo L,et al. Hydrogen Storage by Decalin Dehydrogenation/Naphthalene Hydrogenation Pair over Platinum Catalysts Supported on Activated Carbon[J]. Int J Hydrogen Energy,2008,33(4):1329 - 1334.
[34] Monteiro-Gezork A C A,Natividad R,Winterbottom J M.Hydrogenation of Naphthalene on NiMo-,Ni- and Ru/Al2O3Catalysts:Langmuir-Hinshelwood Kinetic Modeling[J].Catal Today,2008,130(2/4):471 - 485.
[35] Romero C M C,Thybaut J W,Marin G B. Naphthalene Hydrogenation over a NiMo/γ-Al2O3Catalyst:Experimental Study and Kinetic Modeling[J]. Catal Today,2008,130(1):231 - 242.
[36] Kirumakki S R,Shpeizer B G,Sagar G V,et al. Hydrogenation of Naphthalene over NiO/SiO2-Al2O3Catalysts:Structure-Activity Correlation[J]. J Catal,2006,242(2):319 - 331.
[37] 张小菲,邵正锋,毛国强,等. 萘在贵金属Pd、Pt 及Pd-Pt催化剂上的加氢活性及耐硫性能[J] 物理化学学报,2010,26(10):2691 - 2698.
[38] 张坤. 碳纳米纤维负载Pd-Pt催化剂的萘加氢抗硫性能[J].吉林化工学院学报,2008,25(2):8 - 11.
[39] Mandreoli M,Vaccari A,Veggetti E,et al. Vapour Phase Hydrogenation of Naphthalene on a Novel Ni-Containing Mesoporous Aluminosilicate Catalyst[J]. Appl Catal,A,2002,231(1/2):263 - 268.
[40] 王锦惠,李保恩. 萘加氢制四氢萘绝热反应器的研究[J]. 石油化工,1982,11(3):196 - 201,212.
[41] 王智强,李伟,张明慧,等. Ni2Mo3N/Hβ催化剂芳烃饱和加氢与开环性能[J]. 石油学报:石油加工,2005,21(2):23 - 27.
[42] 李洪宝,黄卫国,康小洪,等. 含氮化合物对NiW体系催化剂芳烃加氢性能的影响[J]. 石油炼制与化工,2006,37(10):27 - 31.
[43] 李洪宝,黄卫国,康小洪,等. 载体对Ni2W加氢催化剂活性相及芳烃饱和性能的影响[J]. 石油学报:石油加工,2006,22(6):69 - 75.
[44] Yasuda H,Kameoka T,Sato T,et al. Sulfur-Tolerant Pd-Pt/Al2O3-B2O3Catalysts for Aromatics Hydrogenation[J]. Appl Catal,A,1999,185(2):199 - 201.
[45] Yasuda H,Sato T,Yoshimura Y. Influence of the Acidity of USY Zeolite on the Tolerance of Pd-Pt Catalysts for Aromatics Hydrogenation[J]. Catal Today,1999,50(1):63 - 71.
[46] Rautanen P A,Aittamaa J R,Krause A O I. Liquid Phase Hydrogenation of Tetralin on Ni/Al2O3[J]. Chem Eng Sci,2001,56(4):1247 - 1254.
[47] Mhaouer M,Lemberton J L,Pérot G. Hydrogenation of Tetralin on a Sulfided Ruthenium on KY Zeolite Catalyst:Effect of the Sulfidation Method[J]. Catal Today,1996,29(1/4):241 - 244.
[48] Yasuda H,Higo M,Yoshitomi S,et al. Hydrogenation of Tetralin over Sulfided Nickel-Tungstate/Alumina and Nickel-Molybdate/Alumina Catalysts[J]. Catal Today,1997,39(1/2):77 - 87.
[49] Da Costa P,Lemberton J L,Potvin C,et al. Tetralin Hydrogenation Catalyzed by Mo2C/Al2O3and WC/Al2O3in the Presence of H2S[J]. Catal Today,2001,65(2/4):195 - 200.
[50] 王雷,邱建国,李奉孝. 四氢萘加氢裂化反应动力学[J].石油化工,1999,28(4):240 - 243.
[51] Girgis M J,Gates B C. Reactivities,Reaction Networks,and Kinetics in High-Pressure Catalytic Hydroprocessing[J].Ind Eng Chem Res,1991,30(12):2021 - 2058.
