2,4,6-三{[4-(4-羟乙氧基)苯亚甲胺基]苯乙烯基}均三嗪的结构、光谱及热力学性质
2013-12-17陈自然李为民邓秀华
陈自然,李为民,邓秀华
(1.四川职业技术学院 建筑与环境工程系,四川 遂宁 629000;2.西华师范大学 数学与信息学院,四川 南充 637000)
0 引言
近年来,有机非线性光学(NLO)材料的研究大多集中在构建含电子受体(A),电子给体(D)和π电子共轭的有机分子,如D-π-A,A-π-D-π-A型等有机分子,着力于增大分子的非线性光学系数,改善有机非线性光学材料的光学性能,[1-2]但D-π-A型分子存在效率与透明度的矛盾.[3]为了解决效率与透明度之间的矛盾,Zyss等提出了非偶极非中心对称八极分子(octupolar molecule)作为二阶非线性光学材料的新思想.[4]有关具有三重对称轴的八极分子NLO效应的实验合成研究日益增多,尤其是以吸电子能力较强的均三嗪为核,含有苯乙烯桥侧链三支结构的均三嗪衍生物的合成与光学性能研究,深受化学工作者的关注.尹磊等设计合成了每一支链中均包含两个苯乙烯单位的三支结构的均三嗪衍生物,实验测定其具有显著的双光子吸收协同加强效应.[5]文献[6-7]报道了 2,4,6- 三((4-(4-羟乙氧基)苯亚甲胺基)苯乙烯基)均三嗪(BJZJSQ)等几种含有三重D-π-A结构单元、具有较强二阶非线性效应的均三嗪八极分子,并给出了BJZJSQ分子的红外光谱数据及在DMF溶剂中的最大吸收波长(411.5nm),但未见相关分子结构与性能关系的研究报道.为了能实验合成性能更优异的八极分子非线性光学材料,应用量子化学的理论方法,研究分子结构与性能关系,可为实验合成提供有用的信息.
1 计算方法
杂化密度泛函理论方法和含时密度泛函理论方法被广泛用于分子结构与性质的计算研究.[8]本文使用Gaussian 09程序,采用密度泛函理论B3LYP方法对BJZJSQ分子进行几何结构优化和频率与热力学性质等计算,用含时密度泛函理论TD-B3LYP方法计算电子吸收光谱,并使用PCM模型研究电子光谱的溶剂效应.
2 结果与讨论
2.1 结构优化与分析
在B3LYP/6-31+G*水平上对BJZJSQ分子(气相)的可能构象进行几何优化和频率计算,得到近似平面(存在虚频)与非平面两种构型,分子总能量分别为:-2866.5267082和 2866.53219309a.u.,前者的能量较高,说明非平面构型是最稳定的.图1为BJZJSQ分子最稳定的构型,主要的结构参数见表1.
由图1可知,BJZJSQ分子由一个均三嗪环和A、B、C三个(4-(4-羟乙氧基)苯亚甲胺基)苯乙烯基支链组成.表1中的二面角数据显示,分子中心的2,4,6-三苯乙烯基均三嗪几乎共面,各支链中的乙氧基苯亚甲胺基构成一个面.三支结构中,各侧链中的乙氧基苯亚甲胺基面与苯乙烯基面的二面角约为40°,致使整个分子呈现出三叶扇形结构.计算结果显示,三支分子结构中各分支构型的键长、键角几乎相同,所以表1仅列出了均三嗪环和其中一个分支的主要结构参数.分析A支链的键长可知,均三嗪环与苯乙烯基的C3-C4、C4-C5、C5-C6,苯乙烯基与乙氧基苯亚甲胺基的C12-C13键长都介于C-C单键0.154 nm和C=C双键0.134 nm之间;苯乙烯基与乙氧基苯亚甲胺基的C9-N4、C12-N4键长介于C-N单键0.147 nm和C=N双键0.127 nm之间;苯乙烯基与乙氧基苯亚甲胺基的C16-O1、C19-O1键长介于C-O单键0.143 nm和C=O双键0.122 nm之间,具有一定的双键性质.由此可知,该化合物分子存在较强的π电子共轭效应,表明有较强的电荷离域能力.与文献[6]报道的该化合物具有良好的二阶非线性效应一致.
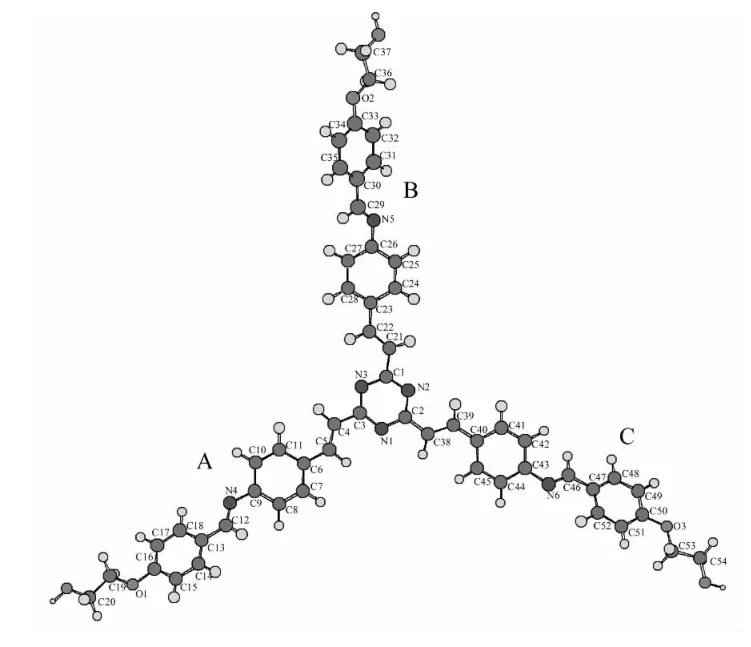
图1 BJZJSQ分子的稳定结构
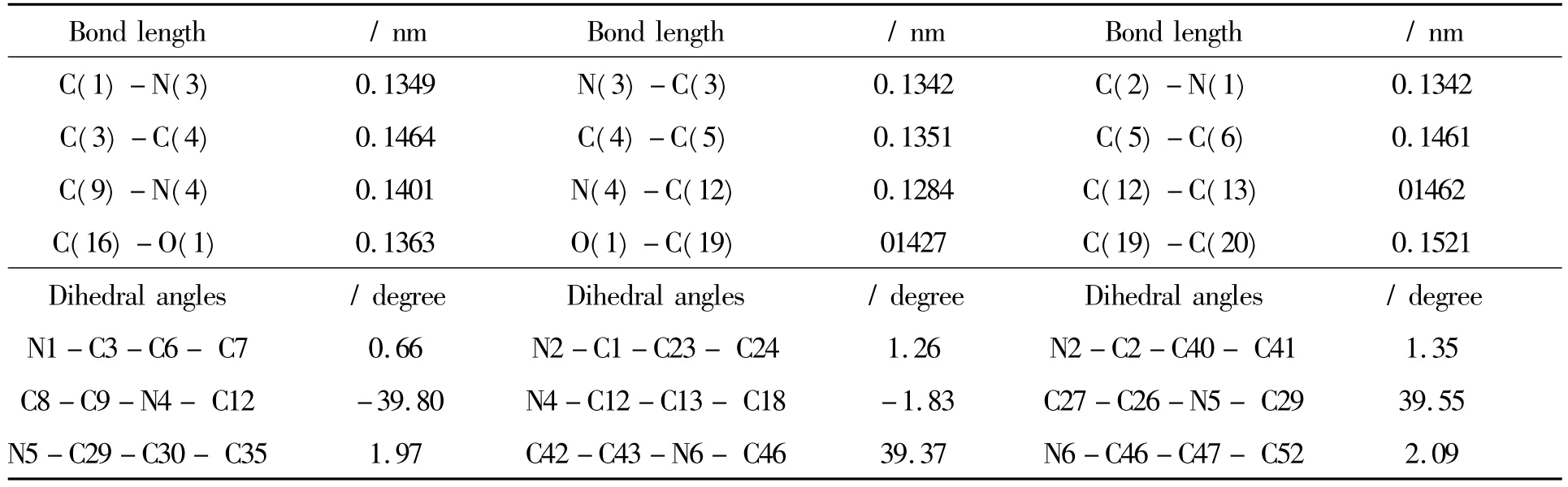
表1 BJZJSQ分子的主要结构参数
2.2 红外光谱数据
表2为BJZJSQ分子的红外光谱(IR)计算数据及实验值.
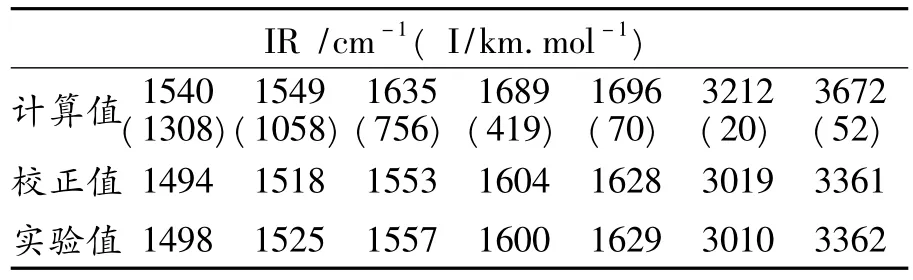
表2 BJZJSQ分子的红外光谱数据
表2数据表明,BJZJSQ分子红外光谱的理论计算值与实验值相差24~310cm-1(0.29~3.92kJ·mol-1),到达化学精度水平要求的误差绝对值在4.2 kJ·mol-1以下.校正因子为0.89~0.91.
2.3 前线分子轨道和电子吸收光谱
分子非线性光学(NLO)性质与基态和激发态的分子轨道组成、电子跃迁及电荷转移等特征相关.在TD-B3LYP/6-31+G*水平上,使用PCM模型的自洽反应场SCRF理论分别计算BJZJSQ分子在苯、乙醇及N,N-二甲基甲酰胺(DMF)溶剂下的紫外吸收光谱,并与气相的紫外吸收光谱相比较以确定其溶剂效应.BJZJSQ分子在DMF溶剂中的前线分子轨道如图2所示.
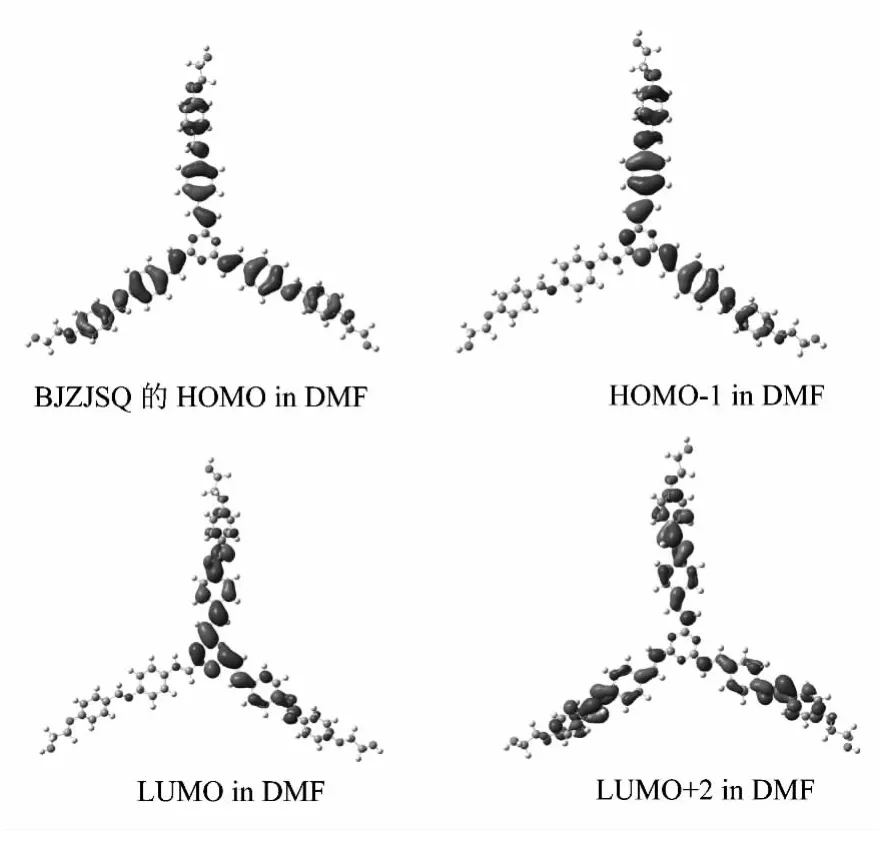
图2 六个分子前线分子轨道
计算得到BJZJSQ分子的HOMO、HOMO-1、HOMO-2、LUMO、LUMO+1及 LUMO+2在气相和不同溶剂相中的电子云分布基本相同,且HOMO-1与HOMO-2电子云分布基本相似,LUMO与LUMO+1电子云分布也近乎相同,所以图3只给出DMF溶液中的HOMO、HOMO-1、LUMO及LUMO+2轨道图.由图3可知,分子的HOMO电子云在三分枝的4-苯亚甲胺基与苯乙烯基两共轭面内平均分布,属于成键π轨道.HOMO-1与HOMO-2电子云主要分布在相邻两分枝的4-苯亚甲胺基与苯乙烯基两共轭面上,属于成键π轨道.LUMO与LUMO+1电子云定域在均三嗪环及其相邻两分枝的苯乙烯基共轭面上,为反键π*轨道.LUMO+2电子云主要定域在三分枝的4-苯亚甲胺基共轭面上,属于反键π*轨道.溶剂对BJZJSQ 分子的 HOMO、HOMO-1、HOMO-2、LUMO、LUMO+1及LUMO+2特征影响不大.
分子吸收光谱数据见表4,吸收光谱图见图3.分析表4数据可知,BJZJSQ分子在气相中的最低能量吸收(也是最强吸收峰)波长位于416 nm,溶剂极性使其红移12~15 nm,均主要对应HOMO→LUMO的π→π*跃迁.在DMF溶剂中的最大吸收波长的计算值431 nm与实验值412nm接近,表明计算结果可信.由图3可知,溶剂相,在355 nm处的弱吸收峰,源于HOMO-2→LUMO+2与HOMO-1→LUMO+2的混合跃迁,对应S0→S8电子跃迁,溶剂极性对其影响不大.
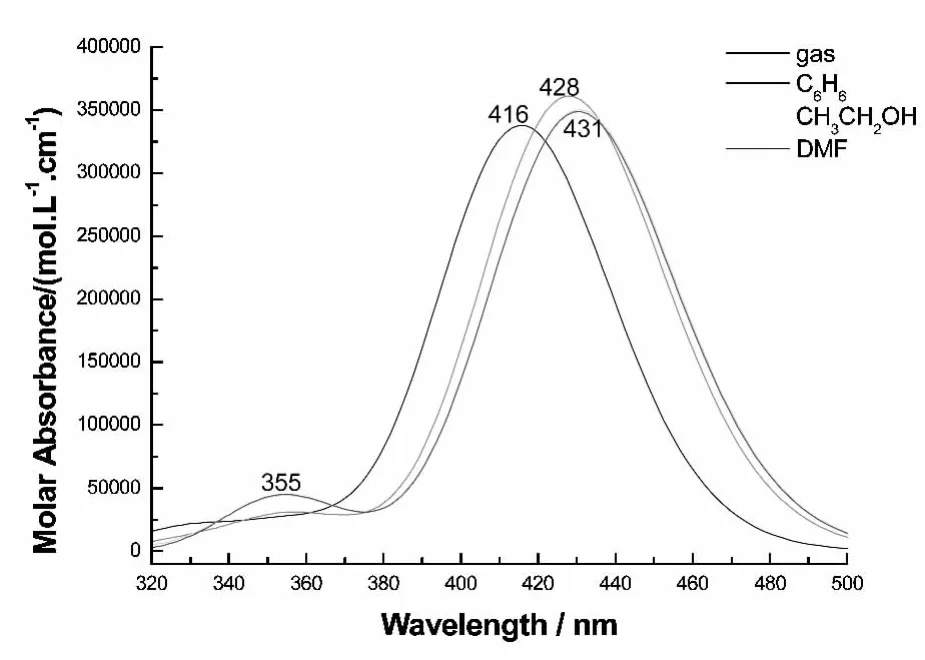
图3 BJZJSQ分子的电子吸收光谱

表3 电子吸收光谱数据
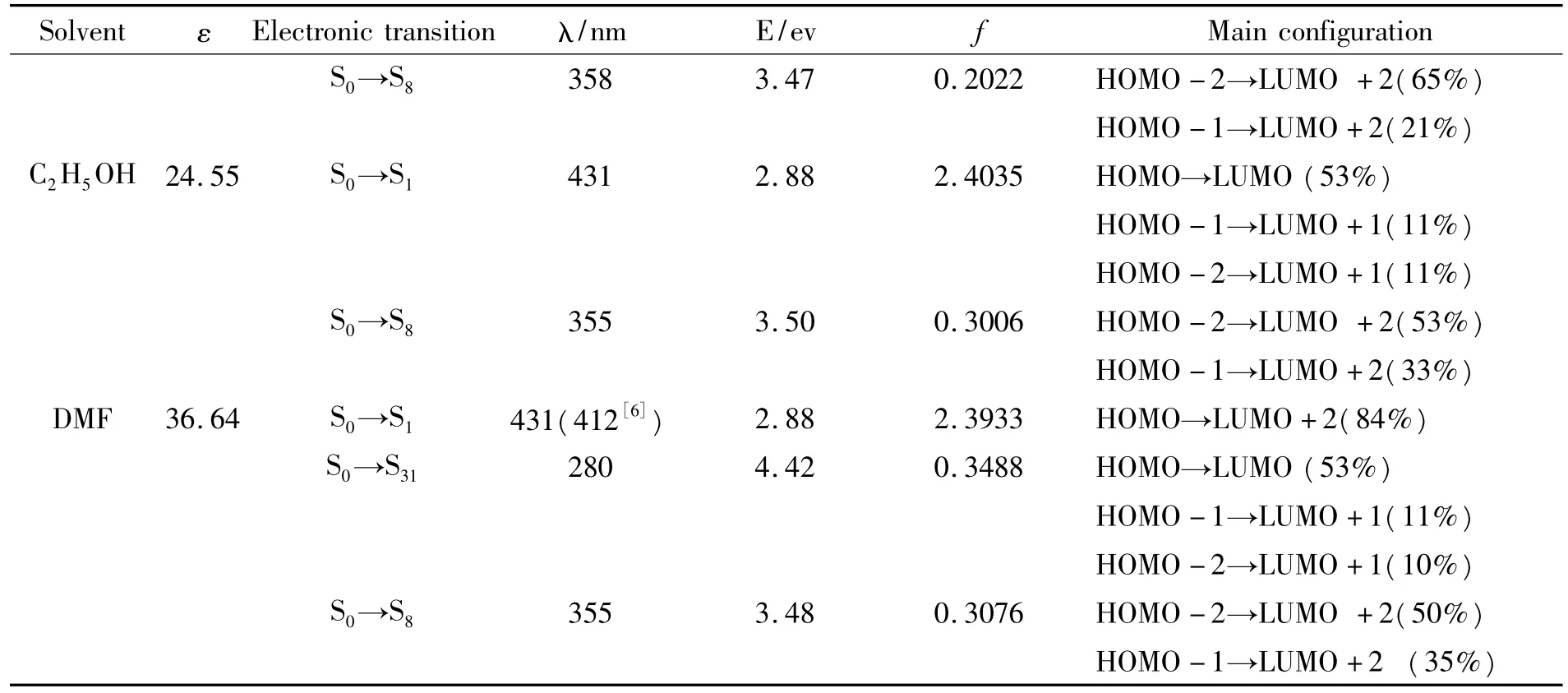
Solvent ε Electronic transition λ/nm E/ev f Main configuration S0→S8 358 3.47 0.2022 HOMO-2→LUMO+2(65%)HOMO-1→LUMO+2(21%)C2H5OH 24.55 S0→S1 431 2.88 2.4035 HOMO→LUMO(53%)HOMO-1→LUMO+1(11%)HOMO-2→LUMO+1(11%)S0→S8 355 3.50 0.3006 HOMO-2→LUMO+2(53%)HOMO-1→LUMO+2(33%)DMF 36.64 S0→S1 431(412[6]) 2.88 2.3933 HOMO→LUMO+2(84%)S0→S31 280 4.42 0.3488 HOMO→LUMO(53%)HOMO-1→LUMO+1(11%)HOMO-2→LUMO+1(10%)S0→S8 355 3.48 0.3076 HOMO-2→LUMO+2(50%)HOMO-1→LUMO+2(35%)
2.4 热力学性质
为了预测BYJXDS分子的标准生成热等热力学性质,对其燃烧反应进行反应热计算.燃烧反应如下:
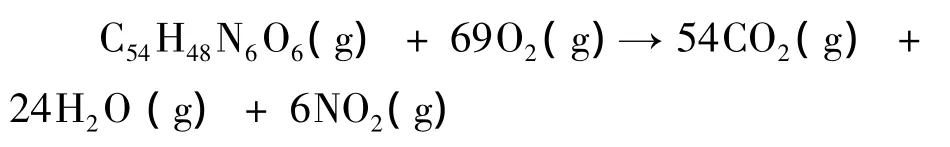
该燃烧反应的标准摩尔反应热△rHm、标准摩尔熵变△rSm、标准摩尔自由能变△rGm的计算公式如下:
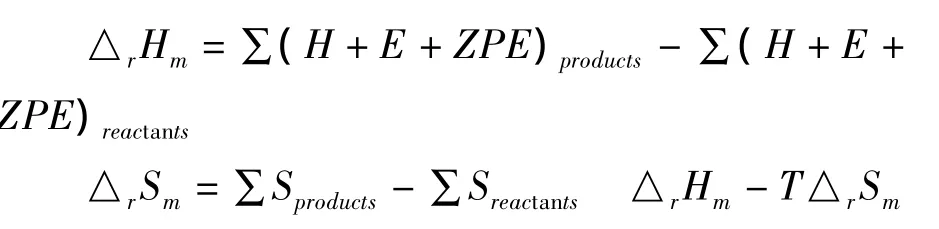
式中E为电子能量,ZPE为零点校正能,H为校正焓,S为熵,G为自由能,products和reactants分别表示所有生成物和反应物.计算得到标题化合物分子的燃烧反应在298.15K时的标准摩尔热力学性质见表4.由资料查到计算所需CO2(g)的△fHm和△fGm分别为-393.51和 -394.38kJ.mol-1,H2O(g)的△fHm和△fGm分别为 -241.83 和 -228.59kJ.mol-1,NO2(g)的△fHm和△fGm分别为 33.85 和 51.84kJ.mol-1.[9]标准摩尔生成焓△fHm和标准摩尔生成自由能△fGm依据反应热的定义按下列公式计算.
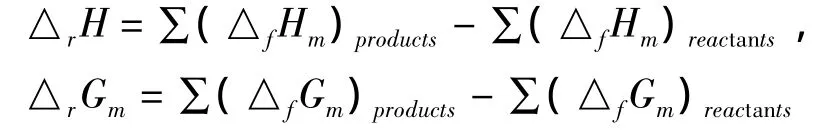
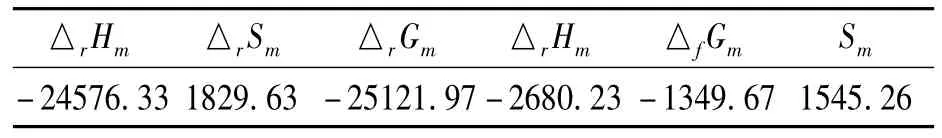
表4 BYJXDS分子的热力学性质
由表4看出,在298.15K、标准大气压下,BJZJSQ化合物分子的燃烧反应是放热、熵增的自发反应,由稳定单质生成BJZJSQ化合物时放热,且能自发进行,这些热力学数据对BJZJSQ化合物分子的其他性质研究提供了参考.
3 结论
采用量子化学计算方法计算得到八极分子2,4,6-三((4-(4-羟乙氧基)苯亚甲胺基)苯乙烯基)均三嗪(BJZJSQ)的结构参数、红外光谱、紫外电子吸收光谱及热力学性质.BJZJSQ分子中心的2,4,6-三苯乙烯基均三嗪几乎在一个平面,各支链中的乙氧基苯亚甲胺基构成另一个平面,整个分子呈现出三叶扇形结构.气相分子的最强吸收峰位于416 nm处,源于HOMO→LUMO、HOMO-1→LUMO及HOMO-2→LUMO+1的混合跃迁,溶剂极性使其红移12—15 nm,溶剂的极性不影响最强吸收峰的跃迁性质.在溶剂相,355 nm处的弱吸收峰对应S0→S8电子跃迁.在298.15 K、标准大气压下,BJZJSQ分子标准摩尔生成焓和标准摩尔生成自由能分别为-2680.23和-1349.67 kJ.mol-1.
[1]陈自然,聂 汉,李 权,等.吡唑啉-噁二唑类有机分子的电子光谱和二阶非线性光学性质[J].化学学报,2011(24):2908-2914.
[2]罗姗姗,仇永清,刘春光,等.六元碳环邻位对称取代的Λ-型分子非线性光学系数的计算[J].高等学校化学学报,2010(7):1436-1442.
[3]Ledoux Zyss J.Organic Molecules for Nonlinear Optics and Photonics[M].Netherlands:Kluwer Academic Publishers,1991:81-103.
[4]Ledoux Zyss J.Nonlinear Optics in Multicolor Media:Theory and Experiments[J].Chem Rev,1994(94):77-105.
[5]尹 磊,方 奇,崔月芝.支型长链均三嗪衍生物的合成与光谱性质[J].化学学报,2005(23):2179-2184.
[6]祁小云,胡泉源,王世敏,等.一种新型八极分子的合成、表征及性能研究[J].化学学报,2012(3):363-366.
[7]祁小云,胡泉源,王世敏,等.二种活性八极分子的合成与表征[J].化学研究与应用,2012(2):296-300.
[8]Chen Z R,Xu Y H,Tao G.Theoretic Study of 3-(4-N-Maleimido)-Phenyl-2,4-dihydro-2H-1,3-benzoxazine-Molecular Structure,Spectrum and Thermodynamic Properties[J].Chinese J Struct Chem,2011(12):1691-1698.
[9]Zhu C Z,Chu Y,Xu H H.Physic Chemistry[M].Beijing:Science Press,2008:556-557.
