安徽省391株结核分枝杆菌临床分离株MLVA基因分型研究
2013-11-16王庆董海燕包训迪赵秀芹徐东芳万康林
王庆 董海燕 包训迪 赵秀芹 徐东芳 万康林
结核病是当今全球范围对人类最具威胁的传染性疾病之一,据世界卫生组织统计显示,全球结核病发病率以每年1%的比率上升,约每年新增患者880万[1]。现代的分子生物学技术与传统的结核流行病学相结合,形成新的结核分子流行病学。多位点可变数目串联重复序列分析(multiple locus variable number tandem repeat analysis,MLVA)就是该学科的一个较典型的范例[2]。MLVA是一种以PCR技术为基础的方法,其操作简单、分辨率高、结果分析容易,具有很高的可重复性,在实验室内和实验室间具有非常好的可比性,并能提供数字化的分型信息,适宜进行大量样本和网络化分析[3]。本研究选取391株结核分枝杆菌临床分离株,采用Supply等[4]新推荐的MLVA(15位点)进行基因分型,以初步了解安徽部分地区结核分枝杆菌的基因型分布及其流行情况,探讨其在今后结核病控制中的应用价值。
材料和方法
一、材料
391株结核病临床分离菌株来自于安徽省胸科医院2011年1—6月的临床痰标本,所有菌株鉴定为结核分枝杆菌,-80℃菌株库保存,标准菌株H37Rv由中国疾病预防控制中心传染病预防控制所提供,作为本实验的对照菌株。
二、方法
1.结核分枝杆菌DNA模板的制备:将结核分枝杆菌临床分离株常规接种于L-J培养基,37℃培养4~8周,至有菌落生长后,取一接种环的菌于400μl TE缓冲液(pH 8.3)中悬菌,100℃煮沸15min,13 059×g离心5min后,上清液备用。
2.PCR扩增:采用25μl PCR反应体系,其中包括2μl模板 DNA、1.5mmol MgCl2,0.2mmol dNTP、上下游引物共30pmol和1.5UTaq DNA酶。分别用15对引物进行PCR扩增。PCR反应条件:预变性94℃10min;循环94℃1min,62℃1min,72℃15min,共40个循环;最后延伸72℃10min。15个位点及引物序列参照文献[5],引物由上海生物工程有限公司合成,详见表1。
3.结果检测:2%琼脂糖凝胶电泳PCR产物,GoldViewTM核酸染料染色,用100bp PCR Marker来确定相对分子质量的大小,以标准菌株H37Rv作为对照,计算出不同结核分枝杆菌各个可变数目串联重复序列(variable number tandem repeats,VNTR)位点的相对分子质量和重复次数。
4.结果分析:先用Gel-Pro analyzer 3.1软件对凝胶进行数字化处理,将指纹图谱数字化后,再用BioNumerics(Version5.0)数据库软件进行聚类分析,将实验菌株进行分型处理。
5.计算 Hunter-Gaston分辨率指数(Hunter-Gaston index,HGI):计算各位点等位基因多样性(h)以及不同VNTR位点组合的HGI,公式如下[6]:
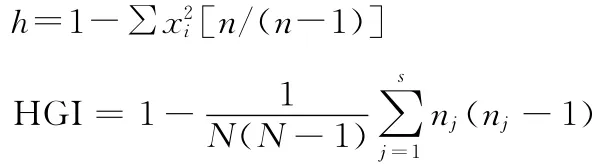
xi为第i等位基因在某个VNTR位点出现的频率,N为实验菌株总数。
N为菌株总数,S是所用分型方法划分的总类型数,nj是属于第j类型的菌株数。
6.计算菌株成簇率:实验结果完全相同的菌株定义为一簇,成簇率为成簇菌株占总试验菌株的百分率。

表1 VNTR位点、重复片段大小及其在H37Rv中的重复次数

续表1
结 果
一、检测标本结果的重复性
阳性对照菌株DNA在同一VNTR位点的扩增片段大小相同。从检测标本中抽取40株菌株,每株不同VNTR位点重复检测3次,每次DNA扩增片段大小完全相符。
二、MLVA多态性检测
本次共选取了15个特异性较好的VNTR位点来进行检测分析,结果显示安徽省结核分枝杆菌的DNA指纹图谱呈现明显的多态性(图1~4),不同位点的多态性即h具有较大差异,其中Mtub21位点显示多态性最高(h为0.591),其次,MIRU26位点h为0.527;ETR-B、ETR-C、ETR-D和 MIRU23显示较低多态性,h 分别为0.125、0.08、0.124和0.137,详见表2。VNTR-15位点 HGI为0.913。
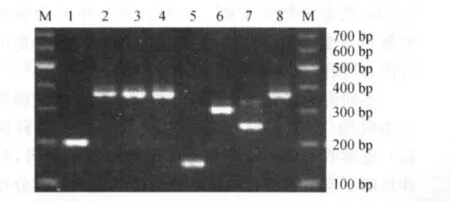
图1 不同菌株在Mtub21位点的PCR产物电泳图
二、MLVA分型
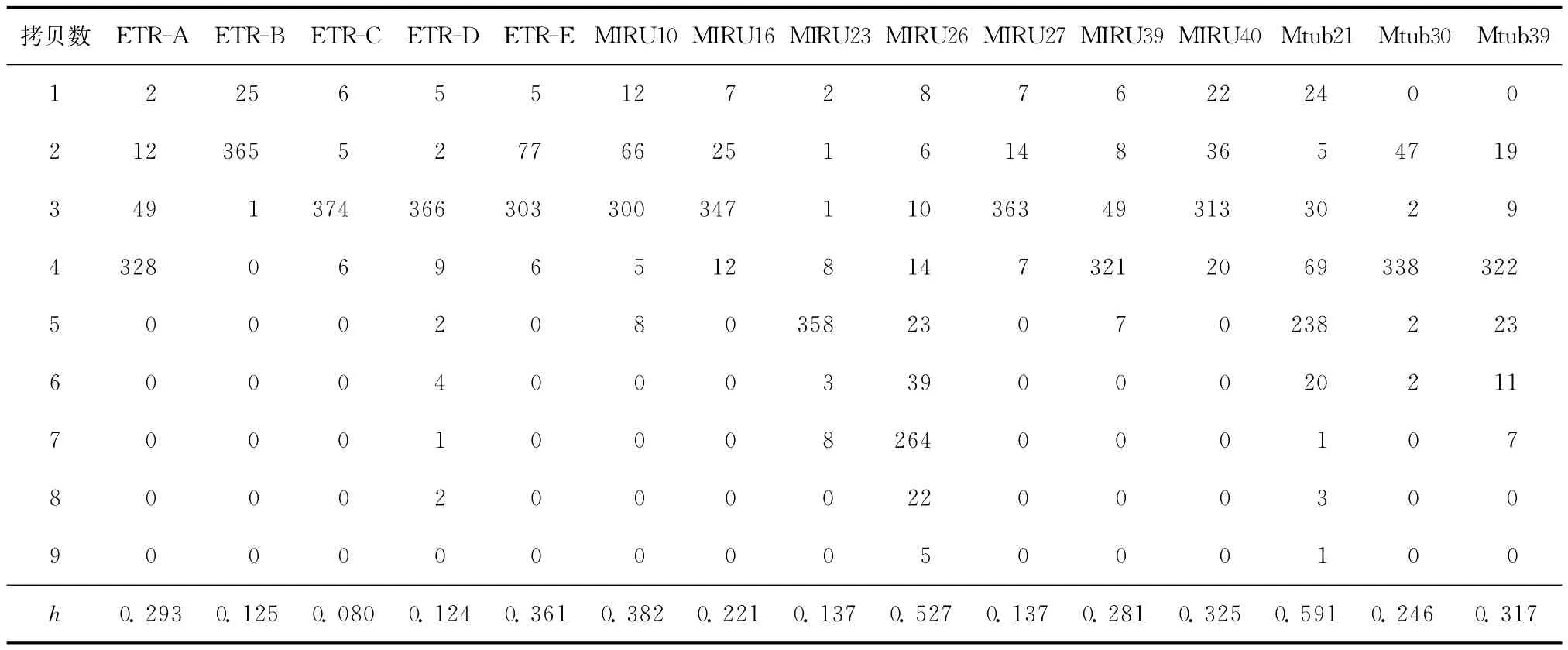
表2 391株结核分枝杆菌15个MLVA位点重复单位多态性分布
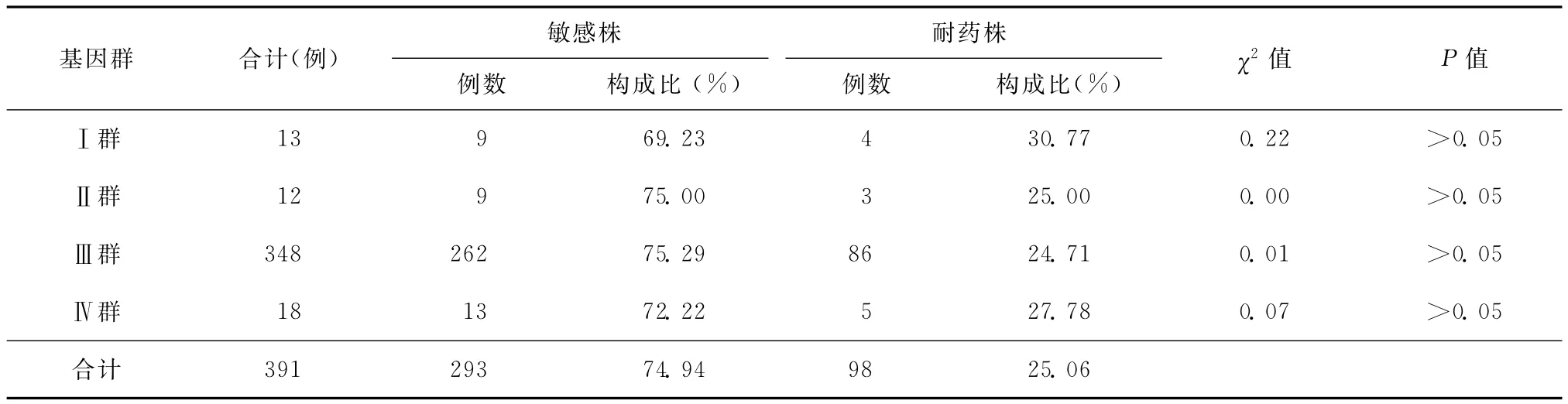
表3 391株结核分枝杆菌药物敏感性和VNTR特征分布关系
1.MLVA分型:经BioNumerics 5.0软件聚类分析,391株分为4个基因群(Ⅰ群、Ⅱ群、Ⅲ群、Ⅳ群),其中Ⅰ群占3.32%(13/391),Ⅱ群占3.07%(12/391),Ⅲ群所占比例最大,为89.00%(348/391),含140个基因型,Ⅳ群占4.60%(18/391);391株菌共含183个基因型,呈明显多态性,其中149株为独特类型,余242株分为34簇,成簇率61.89%(242/391),簇的范围为2~64,最大的一簇包含64株(基因型为424343357333544),占所有菌株的16.4%(64/391)。
2.基因型与耐药性的相关性分析:表3结果显示,391株结核分枝杆菌耐药率占25.06%(98/391),Ⅰ群、Ⅱ群、Ⅲ群、Ⅳ群耐药率分别为30.77%、25.00%、24.71%和27.78%,经卡方检验,基因群分布与药敏间的差异无统计学意义(P值均>0.05)。其中Ⅲ型348株中,利福平耐药率占19.54%(68/348),异烟肼耐药率占17.53%(61/348),同时耐异烟肼与利福平(MDR-TB)的有45株,占12.93%(45/348)。
讨 论
自1998年Frothingham和Meeker-O’Connell首次使用了11个VNTR位点进行基因分型后,近年来许多国家都开展了对这种分型方法的研究,出现了许多种位点的组合方式。Mtb基因组中包含许多类似真核细胞中小卫星序列的散在重复单元(mycobacterial interspersed repetitive unit,MIRU),H37Rv全基因组序列测定结果显示:Mtb基因组中存在41个散在分布的MIRU位点,VNTR位点在Mtb中的分布表现为高度的个体特异性。每个特定的VNTR位点由两部分组成,包括中间的核心区和外围的侧翼区。核心区是由含有至少1个的40~100bp的重复单元组成。一般该重复单元的碱基对数目不变,而串联在一起的重复单元的数目是可变的;外围的侧翼序列具有高度的保守性[7]。
本研究的患者均来源于安徽省法定专业结核病防治机构(安徽省结核病研究所和安徽省胸科医院),连续收集391例痰培养阳性的结核病患者,初步反映了安徽省部分地区结核分枝杆菌的基因分布情况。VNTR位点的等位基因多态性是用于评价各位点对不同菌株的分型能力的指标,就每一个位点而言,它表现为不同菌株在该位点重复序列拷贝数的不同。Hawkey等[8]根据不同位点的等位基因多样性的不同,将其分为3个等级:高分辨能力(h>0.6),中等分辨能力(0.3≤h≤0.6)和低分辨能力(h<0.3)。笔者研究发现VNTR-15位点的总体分辨率是0.913,不同位点之间的多态性差异很大。其中Mtub21、MIRU26具有较高的分辨能力,ETR-E、MIRU10、MIRU40和 Mtub39分辨能力一般,而ETR-B、ETR-C、ETR-D和MIRU23的分辨能力较差。在本研究中其余位点的分辨能力也较差,这与国内外的研究结果并不完全一致[9-11],可能原因有两点:(1)安徽具有某一主要流行菌株,即菌株本身的同源性较高,这对位点分型能力的评估造成了一定程度的影响;(2)本研究中所选择的15个位点尚不足以将所有不同的菌株区分开,提示还应筛选更多的多态性较好的位点组合起来用以区分菌株、提高其分型能力。
391株结核分枝杆菌通过MLVA分型,可分为4个基因群 (Ⅰ群、Ⅱ群、Ⅲ群、Ⅳ群),以Ⅲ群为优势流行菌型,共183个基因型,34个簇,成簇率为61.89%(242/391),最 大 簇 为 64 株 (基 因 型 为424343357333544),说明这34例患者中的每一例患者至少与另外一例患者具有完全相同的基因型,体现了高的成簇率,提示了高度近期传播的可能性。另一方面,可能是笔者采用的分型方法其分辨率不足,未能将有区别的菌株区分开,以致于出现了不精细的成簇,这就需要对更多的位点进行筛选寻找一种最优的位点组合,使得分型结果更趋于真实,最大限度地提高其分型能力。但该结果仍提示应加强对安徽地区主要流行菌株的控制,加速发现传染源,及时切断传播途径,增强易感人群的抵抗力和身体素质,在最大程度上减少传播的发生。如应加强基层单位防治结核的分子流行病学手段,增加特异性位点,对于菌株成簇情况的判定还可能进一步精确,更真实的反映出Mtb的近期传播情况,有利于指导传染源的追踪。本次分析的391株Mtb耐药率占25.06%(98/391),经卡方检验,基因群分布与药敏间的差异无统计学意义(P>0.05)。通过对Ⅲ型菌株的耐药性比较,发现68株对利福平有耐药性,占19.54%(68/348),提示安徽省耐药菌株的监测应重点注意耐利福平菌株的流行趋势。
[1]王庆,杨华,金瑞良,等.结核分枝杆菌重组蛋白Rv0222的表达及血清学鉴定.中国防痨杂志,2012,34(1):36-39.
[2]Malakmadze N,González IM,Oemig T,et al.Unsuspected recent transmission of tuberculosis among high-risk groups:implications of universal tuberculosis genotyping in its detection.Clin Infect Dis,2005,40(3):366-373.
[3] Alonso-Rodríguez N,Martínez-Lirola M,Herránz M,et al.Evaluation of the new advanced 15-loci MIRU-VNTR genotyping tool in Mycobacterium tuberculosis molecular epidemiology studies.BMC Microbiol,2008,8:34.
[4]Supply P,Allix C,Lesjean S,et al.Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis.J Clin Microbiol,2006,44(12):4498-4510.
[5]王晓萌,吕冰,柳正卫,等.Spoligotyping和 MLVA用于71株浙江省结核分枝杆菌临床分离株基因分型的初步研究.中国人兽共患病学报,2008,24(12):1090-1094.
[6]Kam KM,Yip CW,Tse LW,et al.Opitimization of variable number tandem repeat typing set for differentiating Mycobacterium tuberculosis strains in the Beijing family.FEMS Microbiol Lett,2006,256(2):258-265.
[7]Mokrousov I,Ly HM,Otten T,et al.Origin and primary dispersal of the Mycobacterium tuberculosis Beijing genotype:clues from human phylogeogra-phy.Genome Res,2005,15(10):1357-1364.
[8]Hawkey PM,Smith EG,Evans JT,et al.Mycobacterial interspersed repetitive unit typing of Mycobacterium tuberculosis compared to IS6110-based restriction fragment length polymorphism analysis for investigation of apparently clustered cases of tuberculosis.J Clin Microbiol,2003,41(8):3514-3520.
[9]Iwamoto T,Yoshida S,Suzuki K,et al.Hypervariable loci that enhance the discriminatory ability of newly proposed 15-1oci and 24-loci variable-number tandem repeat typing method on Mycobacterium tuberculosis strains predominated by the Bejing family.FEMS Microbiol Lett,2007,270(1):67-74.
[10]Yokoyama E,Kishida K,Uchimura M,et al.Improved differentiation of Mycobacterium tuberculosis strains,including many Beijing genotype strains,using a new combination of variable number of tandem repeats loci.Infect Genet Evol,2007,7(4):499-508.
[11]邓云峰,郑建礼,景辉,等.结核分枝杆菌传播的分子流行病学特征.中华医院感染学杂志,2010,20(10):1380-1383.
