硼酸金属化合物的制备及其电催化活性
2013-11-05刘彦君
刘彦君 金 涛 项 民 刁 鹏
(北京航空航天大学 材料科学与工程学院,北京100191)
随着全球能源需求的日益增长,太阳能作为一种清洁能源,具有分布广泛、容易获得等优点,因此成为一大研究热点.其中,利用特定的催化剂将太阳能转化为化学能或电能,用于分解水制H2和O2,提供了一种储存太阳能的有效方法.20世纪70年代,日本学者首次报道了在光照射条件下,TiO2电极可分解水产生H2[1],掀起了研究热潮.目前,太阳能光解水主要有3种[2]:半导体法,目前研究的半导体已达130多种[3-5],光电化学法[6],光助络合催化法等.
文献[7-12]采用阳极极化法制备了磷酸合钴(CoPi),在室温,pH 7,1标准大气压下即可催化水氧化生成O2.其本身具有很好的稳定性,且可与半导体复合,进一步提高半导体催化性能[13-15].文献[16]在含 Ni2+的 H3BO3/KH2BO3(pH 9.2)中制备了生长厚度可控的硼酸镍(NiBi),在一定的过电势下电催化水分解制氧.
本文在H3BO3/KH2BO3(0.1mol/L,pH 9.2)中电化学沉积制备了硼酸钴(CoBi)、硼酸镍(NiBi)、硼酸锰(MnBi)、硼酸铑(RhBi)、硼酸钯(PdBi)等.对比发现硼酸钴、硼酸镍、硼酸铑具有相对较高的催化活性.探讨了硼酸作为电解质对催化剂制备及催化活性的影响.可进一步研究硼酸金属化合物与半导体复合,并引入光照,从而在低成本、高可操作性条件下实现太阳能的储存与应用.
1 实验部分
1.1 试剂和仪器
硼酸(H3BO3),氯化钴(CoCl2·6H2O),醋酸镍 (Ni(CH3COO)2·3H2O),无水醋酸锰(Mn(CH3COO)2),三氯化铑(RhCl3·3H2O),氯钯酸钠(Na2PdCl4),氢氧化钾(KOH).所有药品均为分析纯.所有溶液由高纯水配置.
CHI660A电化学工作站(CH Instruments Co.).电化学反应采用三电极体系.其中,饱和甘汞电极(SCE,Saturated Calomel Electrode))作参比电极,大面积的铂电极作辅助电极.本文所有的电极电位均以 SCE作参比.场发射扫描电镜(SEM,Hitachi,S-4800)和 XRD 衍射仪(Rigaku,Rint2000,Cu Ka)对硼酸金属化合物形貌与结构进行分析.
1.2 实验过程
1.2.1 硼酸金属化合物薄膜的制备
配制氯化钴(CoCl2·6H2O)、醋酸镍(Ni(CH3COO)2·3H2O)、无水醋酸锰(Mn(CH3COO)2)、三氯化铑(RhCl3·3H2O)、氯钯酸钠(Na2PdCl4)的水溶液,均为5 mmol/L.H3BO3/KH2BO3(0.1 mol/L,pH 9.2)作为支持电解质,氧化铟锡(ITO,Indium Tin Oxide)玻璃为工作电极.在电位窗口0~1.4 V下进行循环伏安扫描,确定相应的硼酸金属化合物生长的特定电位.采用恒电位法沉积硼酸金属化合物薄膜.样品洗净干燥后进行SEM,XRD表征.
1.2.2 硼酸金属化合物薄膜催化水氧化制氧
沉积有化合物的ITO为工作电极,H3BO3/KH2BO3缓冲溶液(0.1 mol/L,pH 9.2)作支持电解质.在电位窗口0.2~1.4 V下进行线性扫描,记录电流随电位升高的变化情况.
2 结果与讨论
2.1 硼酸金属化合物薄膜的制备
图1a~图1d为ITO分别在含有0.5 mmol/L的 Co2+,Ni2+,Mn2+,Rh3+的 H3BO3/KH2BO3(0.1mol/L,pH 9.2)缓冲溶液中的循环伏安(CV)曲线,图1e为Pd2+在H3BO3/KH2BO3缓冲溶液(pH 7.2)中的CV曲线.
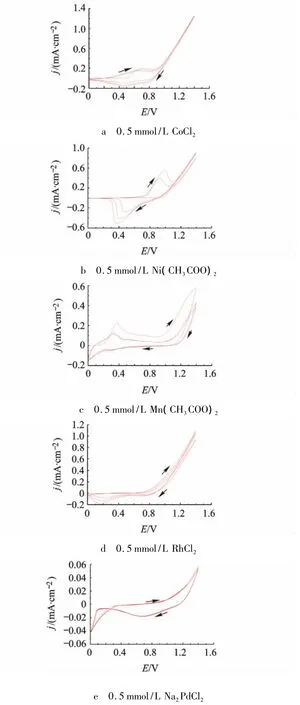
图1 ITO在含对应金属离子的H3BO3/KH2BO3缓冲溶液(pH 9.2)中的CV曲线(0.1 V/s)
由图1a可得在阳极氧化过程中,0.5 V左右Co2+开始被氧化,0.6 V出现氧化峰,阴极还原过程中,0.45 V出现Co2+氧化产物的还原峰.图1b阳极氧化过程,0.9 V为Ni2+氧化峰,且随扫描次数增多氧化峰正移,氧化电流逐渐增大.图1c第1圈扫描0.4 V出现Mn2+的氧化峰,随扫描次数增多,氧化峰稳定在0.35 V,氧化电流比第1圈正扫减小.图1d中Rh3+在0.8 V左右被氧化,其氧化峰与水的氧化峰未分离,氧化电流均随扫描次数增多而增大,说明氧化产物逐渐增多.0.2 V出现Rh3+氧化产物的还原峰.图1e中Pd2+在1.0V左右开始被氧化,0.7 V出现一较小的Pd2+氧化产物的还原峰.由以上分析选择 Co2+,Ni2+,Mn2+,Rh3+,Pd2+的沉积电位分别为:0.6 V,0.9 V,0.35 V,0.8 V,1.0 V.
由图2可知,CoBi为纳米球状(图2a),粒子尺寸较大.NiBi为纳米网状(图2b).MnBi由微小粒子聚集成连续的厚度不均匀的薄膜,部分区域粒子聚集成较大的块体,即图中较亮的区域 (图2c).RhBi由纳米球颗粒聚集成连续薄膜(图2d),部分区域破裂.PdBi由纳米球状聚集成致密的均匀的薄膜,并有较大尺寸的颗粒镶嵌在膜上(图2e).XRD 图中 30°,35°,50°,60°4 个衍射峰均为ITO的衍射峰,说明几种硼酸金属化合物均为无定形结构.

图2 不同硼酸金属化合物的SEM图和XRD图
2.2 不同硼酸金属化合物催化水氧化制氧活性
采用阳极氧化沉积法分别制备了CoBi,NiBi,MnBi,RhBi,PdBi几种薄膜.在 0.1 mol/L 的H3BO3/KH2BO3缓冲溶液中进行线性电势扫描测试,以对比其催化水分解制氧活性,结果如图3所示.CoBi,NiBi和RhBi体系中起峰电势比PdBi体系的更负,而MnBi体系尽管起峰电势较负,但在1.0 V左右有一很宽的氧化峰,这可能是MnBi被氧化而不是H2O的氧化.
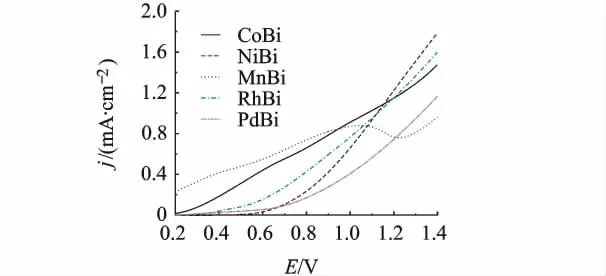
图3 不同硼酸金属化合物在H3BO3/KH2BO3
反应式(1)的标准电极电势ψθ=1.23V(相对于NHE(标准氢电极)),相对饱和甘汞电极(SCE)电极为1 V,故选择比较E>1 V时同一电位下电流大小.不同催化剂体系电位1.2 V时电流响应曲线,如图4所示.CoBi,NiBi和RhBi体系中氧化电流达到0.75 mA/cm2,而 MnBi和 PdBi体系中氧化电流很小,分别为 0.6 μA/cm2和0.1 μA/cm2.故由起峰电势位置和同一电位下电流大小得出CoBi,NiBi和RhBi具有较高的催化活性,而MnBi和PdBi催化性能较低.
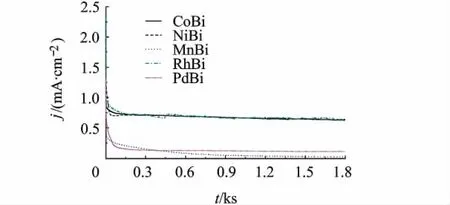
图4 H3BO3/KH2BO3(0.1 mol/L,pH 9.2)缓冲溶液中不同硼酸金属化合物在恒定电位1.2 V的电流响应曲线
2.3 硼酸盐缓冲溶液对硼酸金属化合物催化水氧化制氧的影响
对比0.5 mmol/L CoCl2在0.1 mol/L Na2SO4溶液和0.1 mol/L H3BO3/KH2BO3缓冲溶液中的CV曲线(如图5所示).在硼酸盐缓冲溶液中,正扫0.60 V出现Co2+的氧化峰,Na2SO4溶液中没有出现Co2+单独的氧化峰,而是与水的氧化峰重叠.说明在硼酸盐缓冲溶液中Co2+更易被氧化,因而有利于形成CoBi.
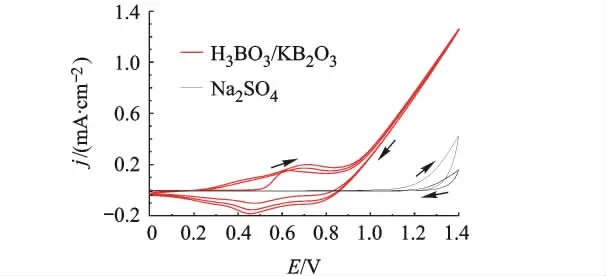
图5 0.5 mol/L CoCl2在0.1 mol/L H3BO3/KH2BO3缓冲溶液和0.1mol/L Na2SO4中的CV图(0.1 V/s)
2.4 pH值对硼酸金属化合物催化水氧化制氧活性的影响
研究同一条件下制备的CoBi在不同pH值的H3BO3/KH2BO3溶液中催化水分解活性,见图6.同一电流密度下氧化电位随电解质溶液pH值的增大而负移.且在水的氧化之前,有一氧化峰随pH值增大逐渐明显并负移.说明溶液中有其他氧化反应进行,且氧化电位随pH值增大而减小.
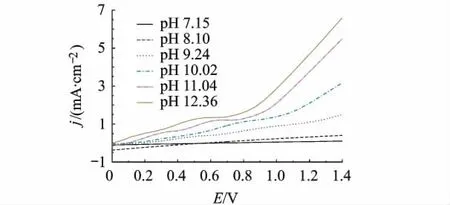
图6 CoBi在不同pH值的H3BO3/KH2BO3(0.1 mol/L)缓冲溶液中的线性电势扫描图(0.1 V/s)
对于反应式(1),由能斯特方程 ψ=ψθ-0.059 pH可知,氧化电位随pH值增大而减小.为进一步研究图6中现象是否符合能斯特方程的变化,对比恒电流下CoBi催化水氧化电位与标准电极电势随pH值变化情况(图7).发现pH值在7~11 范围内,H3BO3/KH2BO3中∂E/∂pH >-59 mV,即CoBi催化水氧化电位随pH值下降速率要比理论值大很多,说明图6中电位随pH值降低不仅仅是能斯特方程影响的结果.pH>11时,∂E/∂pH接近-59 mV,即此时电位随pH值增大而减小的情况接近能斯特方程的结果.
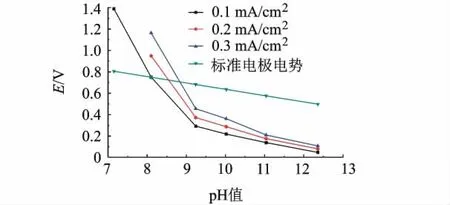
图7 CoBi在H3BO3/KH2BO3(0.1 mol/L)缓冲溶液中不同电流密度下电位随pH值变化关系图
图8显示了H3BO3存在形态随pH值变化情况.pH值在7~11范围内,随pH值增大溶液中H3BO3逐渐减少,而H2BO3-和逐渐增多,即电解质溶液接受质子能力逐渐增强,电解质溶液结合更多的质子,有利于反应式(1)向右进行,从而进一步降低了反应式(1)的氧化还原电位.
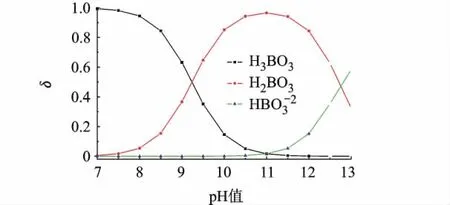
图8 H3BO3/KH2BO3缓冲溶液中H3BO3存在形态随pH值变化关系图
由此提出以下假设:如图9所示,在H3BO3/KH2BO3缓冲溶液中,硼酸金属化合物催化水氧化制氧,H2BO-3接受质子形成H3BO3,从而促使氧气不断生成.而硼酸金属化合物被还原,在一定电位下,催化剂又被氧化,实现自我修复,从而可以重复利用.
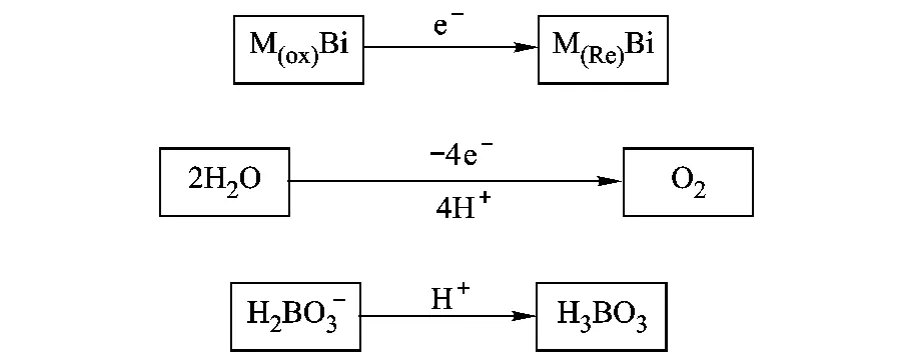
图9 硼酸金属化合物在H3BO3/KH2BO3溶液中催化水分解原理图
3 结论
采用电化学沉积法在ITO表面制备了CoBi,NiBi,MnBi,RhBi,PdBi 几种硼酸金属化合物薄膜.对其形貌和结构进行表征表明几种薄膜均为无定形结构.比较其催化水氧化制氧性能发现,CoBi,NiBi,RhBi具有较高的催化活性,而 MnBi,PdBi催化活性较低.硼酸电解质与其他电解质相比是很好的质子接受体,有利于催化剂的形成和硼酸金属化合物催化水氧化制氧过程的进行.pH值在7~11范围内,随着 pH值增大,溶液中H2BO3-和逐渐增多,能结合更多的质子,从而有利于水氧化的进行.在催化水氧化制氧过程中催化剂被还原,硼酸电解质为催化剂提供质子将其氧化,由此实现了催化剂的循环利用.
References)
[1] Fujishima A,Honda K.Electrochemical photolysis of water at a semiconductor electrode[J].Nature,1972,238(5358):37-38
[2]陈启元,兰可尹,周澜.半导体光解水研究进展[J].材料导报,2005,19(1):20-23 Chen Qiyuan,Lan Keyin,Zhou Lan.Progress on semiconductor photocatalysts for water decomposition[J].Materials Review,2005,19(1):20-23(in Chinese)
[3] Osterloh F E.Inorganic materials as catalysts for photochemical splitting of water[J].Chem Mater,2008,20(1):35-54
[4] Kazuhiko M,Kazunari D.Meeting the clean energy demand:nanostructure architectures for solar energy conversion[J].J Phys Chem C,2007,111(7):7851-7861
[5] BardA J,Fox M A.Artificial photosynthesis:solar splitting of water to hydrogen and oxygen[J].Acc Chem Res,1995,28(3):141-145
[6] Sun Jianwei,Zhong D K,Gamelin D R.Composite photoanodes for photoelectrochemical solar water splitting[J].Energy Environ Sci,2010,3:1252-1261
[7] Surendranath Y,Kanan M W,Nocera D G.Mechanistic studies of the oxygen evolution reaction by a cobalt-phosphate catalyst at neutral pH[J].J Am Chem Soc,2010,132(46):16501-16509
[8]Kanan M W,Nocera D G.In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+[J].Science,2008,321(5892):1072-1075
[9] Surendranath Y,Dincǎ M,Nocera D G.Electrolyte-dependent electrosynthesis and activity of cobalt-based water oxidation catalysts[J].J Am Chem Soc,2009,131(7):2615-2620
[10] Lutterman D A,Surendranath Y,Nocera D G.A self-healing oxygen-evolving catalyst[J].J Am Chem Soc,2009,131(11):3838-3839
[11] Kanan M W,Yano J,Surendranath Y,et.al.Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy[J].J Am Chem Soc,2010,132(39):13692-13701
[12] Mcalpin J G,Surendranath Y,Dinca M,et al.EPR evidence for Co(IV)species produced during water oxidation at neutral pH[J].J Am Chem Soc,2010,132(20):6882-6883
[13] Zhong D K,Gamelin D R.Photoelectrochemical water oxidation by cobalt catalyst(“Co-Pi”)/α-Fe2O3composite photoanodes:oxygen evolution and resolution of a kinetic bottleneck[J].J Am Chem Soc,2010,132(12):4202-4207
[14] Zhong D K,Sun Jianwei,Inumaru H,et al.Solar water oxidation by composite catalyst/α-Fe2O3photoanodes[J].J Am Chem Soc,2009,131(17):6086-6087
[15] Steinmiller E M P,Choi K S.Photochemical deposition of cobalt-based oxygen evolving catalyst on a semiconductor photoanode for solar oxygen production[J].Proc Natl Acad Sci,2009,106(49):20633-20636
[16] Dincǎ M,Surendranath Y,Nocera D G.Nickel-borate oxygen-evolving catalyst that functions under benign conditions[J].Proc Natl Acad Sci,2010,107(23):10337-10341
