Acetonitrile (CH3CN) and methyl isocyanide(CH3NC) adsorption on Pt(111) surface: a DFT study
2013-11-01HANXinyan韩新艳RENJunCAODuanlin曹端林ZHUJiaping朱佳平
HAN Xin-yan (韩新艳), REN Jun (任 君), CAO Duan-lin (曹端林), ZHU Jia-ping (朱佳平)
(College of Chemical Engineering and environment, North Universtiy of China, Taiyuan 030051, China)
Acetonitrile (CH3CN) and methyl isocyanide(CH3NC) adsorption on Pt(111) surface: a DFT study
HAN Xin-yan (韩新艳), REN Jun (任 君), CAO Duan-lin (曹端林), ZHU Jia-ping (朱佳平)
(College of Chemical Engineering and environment, North Universtiy of China, Taiyuan 030051, China)
The adsorption of CH3CN and CH3NC on the Pt(111) surface at the 1/4 monolayer (ML) coverage has been carried out at the level of density functional theory for understanding hydrogenation processes of nitriles. The most favored adsorption structure for CH3CN is the C—N bond almost parallel to the surface with the C—N bond interaction with adjacent surface Pt atoms. For CH3NC, the most stable configuration is the CH3NC locates at the face center cubic (fcc) site with the C-atom bonded to three Pt atoms. In addition, the HCN and HNC adsorption has been computed, and the adsorption pattern is nearly similar to the CH3CN and CH3NC, respectively. The adsorbed molecules rehybridize on the surface, becoming non-linear with a bent C—C—N or C—N—C angle. Furthermore, the binding mechanism of these molecules on the Pt(111) surface is also analyzed.
acetonitrile; methyl isocyanide; adsorption; Pt(111) surface; density functional theory(DFT)
Acetonitrile (CH3CN) and its isomer, methyl isocyanide (CH3NC), have been relatively often used for determining geometrical structure and bonding configuration on single crystal metal surfaces[9-17]. Their application is based on the fact that these compounds are isoelectronic with CO (the most frequently applied probe molecule). While CO generally bonds on Group VIII metals via a synergistic bonding scheme with both donation from the metal into empty 2π*orbitals on CO and donation from the filled 6σ orbital of CO to the metal, methyl isocyanide is a significantly weaker π-acid since its 2π*level lies much higher in energy than that of CO. Therefore, it is interesting, from a fundamental point of view, to compare the surface chemistry of these molecules. In addition, heterogeneous catalytic transformation of acetonitrile has rarely been investigated. Experimental studies on the ultra violet (UV) irradiation of CH3CN adsorbed on TiO2in the presence of oxygen indicated[10]the formation of H2O, CO2, surface CO3and surface isocyanate (NCO). The formation of surface CH3CONH2, η2(N,O)—CH3CONH, CH3COO(a), HCOO(a), NCO(a) and CN-containing species was observed, when the UV irradiation of CH3CN adsorbed on TiO2was performed in the absence of oxygen[11].
Isocyanide adsorption has been studied previously on various metal surfaces. On Pt(111) surface[12,14], CH3CN was found to adsorb weakly to the surface in a parallel structure (through both the terminal carbon and the nitrogen), and CH3NC was found to bond strongly in an upright structure with the terminal carbon bonded to two metal atoms. Acetonitrile (methyl cyanide) was investigated further on Pt(111)[13]and again found to adsorb parallel to the surface with a possible interaction between the β-hydrogens and the surface. The conclusion that CN axis is parallel to the bridge and the adsorption energy is the largest are also calculated by Alexis Markovits and Christian Minot[14].On supported Pt/SiO2, similar results were found for both compounds. On powdered Au, isocyanides were bound weakly to the metal in a linear structure with the terminal carbon bonded to one metal atom. Isocyanides were found to adsorb strongly on Ni(111) and Ni(100) in a parallel structure[15-17]. On Rh(111) surface[16], the isocyanide bonds strongly also in a parallel structure at low coverage. On supported Rh/Al2O3[17], the isocyanide was found to stand up again. Finally, on Ag(311)[18], isocyanides bond wealy in a parallel or close to parallel orientation. So, the adsorption of cyanide on the metal surface has not been investigated systematically.
Quantum chemical methods have become new tools for investigating the structure of active surfaces and determining reaction mechanisms. With recent development, density functional theory (DFT) is capable of providing qualitative and, in many cases, quantitative insights into surface science and catalysis. In this paper, we report a systematic DFT study on CH3CN and CH3NC adsorption on Pt(111) in order to get insight into their surfaces and structures. Furthermore, the aim of the present work is to determine the surface species formed on Pt(111) and to detect the gas products during the interaction of acetonitrile with noble metal catalysts. This study would be helpful to find an effective catalyst in breaking of C—N bond of cyanide compounds.
1 Methods and models
All calculation are done with the Cambridge sequential total energy package[19]. DFT calculation within the generalized gradient approximation (GGA) using the Perdew, Burke and Ernzerhof (PBE) functional[20]was carried out to study nitromethane adsorption on the surfaces of Pt(111). Ionic cores were described by the ultrasoft pseudopotential[21], and the Kohn-Sham one-electron states were expanded in a plane wave basis set up to 340 eV. The difference of the adsorption energy at the level of cutoff between 340 and 360 eV was about 0.02 eV. A Fermi smearing of 0.1 eV was utilized. Brillouin zone integration was approximated by a sum over special k-points chosen using the Monkhorst-Pack scheme[22]. A spin-restricted approach was used for clean surface models since polarization effects were found to be negligible. The convergence criteria for structure optimization and energy calculation were set to (a) self-consisten field (SCF) tolerance of 2.0 ×10-6eV/atom, (b) energy tolerance of 2.0 ×10-5eV/atom, (c) maximum force tolerance of 0.05 eV/-, and (d) maximum displacement tolerance of 2.0 ×10-3-. A4× 4 × 1 k-grid sampling within the Brillouin zones was used in the p(2×2) unit cell and a vacuum region of 15 -. We also tested the k-point sampling by using the 5×5×1 Monkhorst-Pack meshes for the unit cell, and the change in energy is less than 0.03 eV. Although the PBE functional can give reliable optimized geometry, it tends to overbind adsorbate on metal surface[23,24]. We further carried out the RPBE single point energy calculations on the PBE optimized geometries and use the RPBE energy for discussion.
In order to describe the interaction between adsorbates and Pt-slab, we defined the adsorption energy as

where E(slab), E(adsorbates) and E(adsorbates / slab) are the total energy of the optimized slab of the surface, gas-phase adsorbate and adsorbate-slab complex, respectively.
2 Results and discussion
2.1 CH3CN adsorption on Pt(111) surface
Initially, we checked that the properties of the isolated CH3CN were accurately reproduced. The calculated gas phase C—N and C—C bond lengths is 1.169 and 1.435 -, respectively. The C—C—N angle is 179.5 - in good agreement with experiment value (C—N:1.158 -, C—C: 1.460 -, and C—C—N: 180.0 -). The Pt(111) surface exhibits four high-symmetry adsorption sites as shown in Fig.1. The adsorbed species could be located on top, bridge, face cerder cubic kinds of (fcc) hollow or hexagonal close-packed (hcp) hollow sites, hereafter labeled t, b, f and h, respectively. We designated all possible adsorption structures for CH3CN on the surface, top, bridge, fcc hollow or hcp hollow, based upon the position of the N atom, only three relatively stable configuration were obtained in Fig.1. The calculated adsorption energies and structural parameters for CH3CN are listed in Table 1. In I (2-fold bridge site), CH3CN interacts with two adjacent Pt atoms via the C—N bond and forms one Pt—N and one Pt—C bond and the C—N axis almost parallel to the surface. The C—N bond is elongated compared to free CH3CN (1.252 vs 1.15 -) (Fig.1). The C—C bond is stretched to 1.474- corresponding to gas phase calculated value 1.435 -, and the C—C—N bond angle is reduced to 129.1 -. The length of Pt—N and Pt—C bonds is 2.032 and 2.053 -, respectively. These results are close to those reported for the acetonitrile adsorption on Ni(111)[25], which showed that a rehybridization of the acetonitrile molecule into a sp2 configuration. In II (top1 site), the acetonitrile molecule linearly adsorbed on the surface by one C atom atop on one Pt and it bonded to the surface only by the nitrogen atom with the C—N axis almost perpendicular to the surface. The C—N bond length is about 1.166 - and the C—C—N bond angle is 178.7 -. For the bridge adsorption, each of the possible bridge adsorption sites on the surface was considered, but the acetonitrile molecule adsorption configuration changed from bridge to top site during optimization (III, top2 site). The C—C bond length is about 1.667-. As given in Table 1, I is the most stable adsorbed form with the largest adsorption energy (-0.48 eV), while II and III have lower adsorption energies (-0.33 and -0.30 eV).
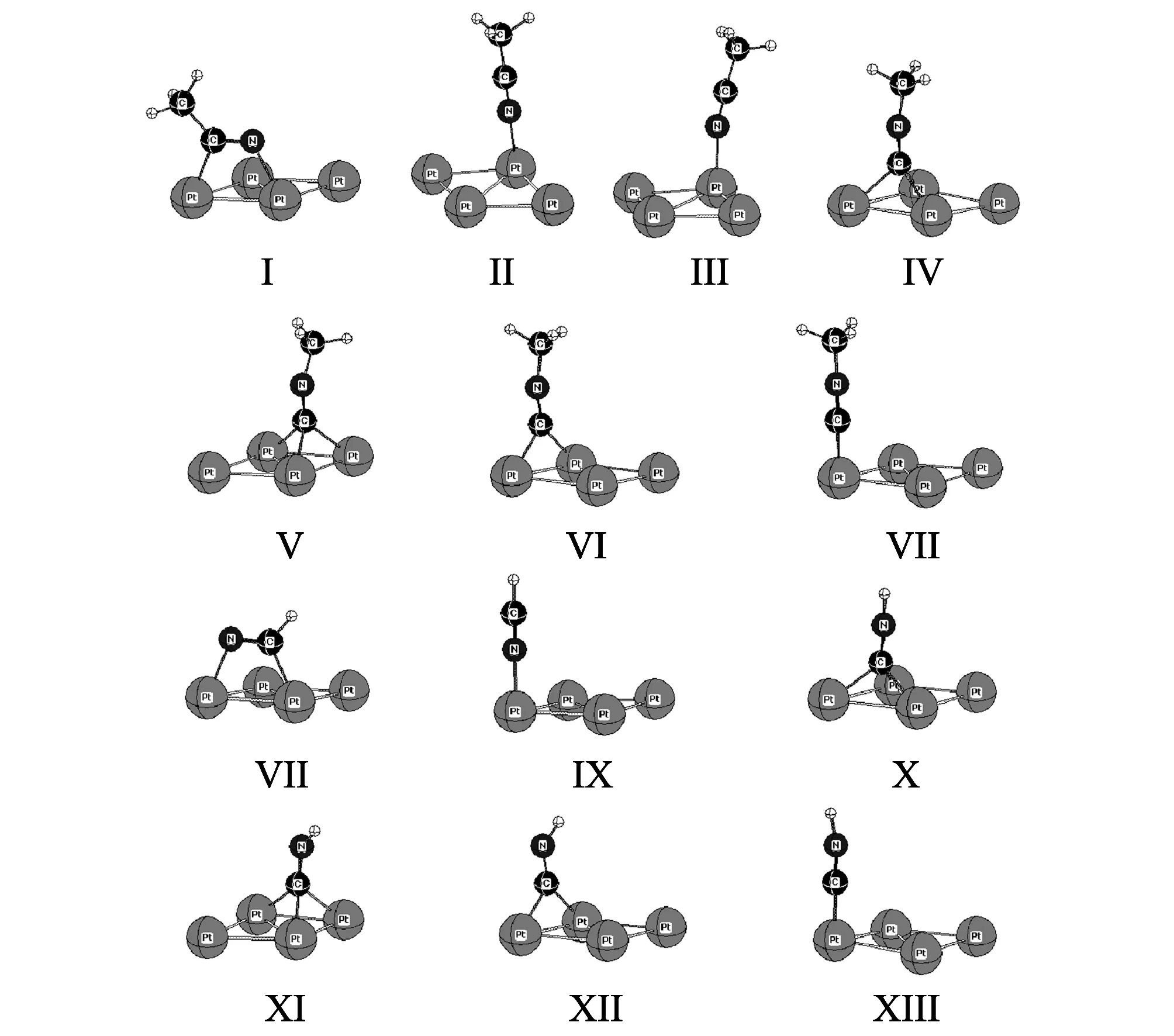
Fig.1 Adsorption of CH3CN, CH3NC, HCN and HNC on Pt(111) surface at 1/4 ML

Table 1 Calculated adsorption energies (Eads, eV) and structural parameters (d, - and θ, deg) for CH3CN, CH3NC, HCN and HNC adsorption on the Pt(111)-2×2 surfaces
2.2 CH3NC adsorption on Pt(111) surface
The interaction of isocyanides with transition metals has been extensively studied in the context of organometallic chemistry. Recently, scanning tunneling microscopy(STM)[26]and reflection absorption infrared spectroscopy and temperature-programmed desorption(TPD)[27]were used to investigate the adsorption of methyl isocyanide (CNCH3) on the Pt(111) surface and its reaction to form methylaminocarbyne(CNHCH3). It indicated that methyl isocyanide is found to adsorb at on-topsites at low coverage and at both on-top and 2-fold bridge sites at higher coverage. In this section, we have mainly focused on the structure of the adsorbed molecule and the adsorption site. For CH3NC adsorption, Fig.1 shows four adsorption forms (IV, V, VI and VII). In IV, the CH3NC locates at the fcc site with the C-atom bonded to three Pt atoms and the Pt—C bond lengths are 2.109, 2.116 and 2.135, respectively. The C—C—N bond angle is 178.9 - and the two C—N bond lengths are 1.210 and 1.389 -, respectively. In V, the CH3NC is situated at hcp site by the C-atom adsorption and the length of the Pt—C bond is changed from 2.101 to 2.125 -. The C—N bond is stretched about 0.044 and 0.22 -, respectively. In V, the CH3NC adsorbed at the hcp site with the C-atom interaction with three Pt atoms on the surface with three Pt—C distances of 2.101, 2.125 and 2.115 -. The two C—N bond lengths are extended to 1.213 and 1.387 -, respectively. The C—C—N bond angle is 162.1 -. In VI, the carbon of the CH3NC bridges on two Pt atoms with Pt—C bond lengths of 2.042 and 2.031 - and the C—N distances of 1.393 and 1.201 -. In IV, the carbon of the CH3NC atop on one Pt atom with the C—N distances of 1.397 and 1.177 -. In Table 1, IV is the most stable adsorption mode (-1.57 eV), followed by V(-1.55 eV), while VI and IV are much less stable (-1.50 and -1.48 eV). It is interesting to note that the CH3NC adsorption on hcp site is similar to the fcc site due to the very small of the energy difference (-0.02 eV). Considering a possible equilibrium between the two states, the chance of the CH3NC adsorption on fcc and hcp sites is same thermodynamically. On this basis, the CH3NC should dissociate dominantly and directly on the two sites, while the bridge and top sites play a minor role.
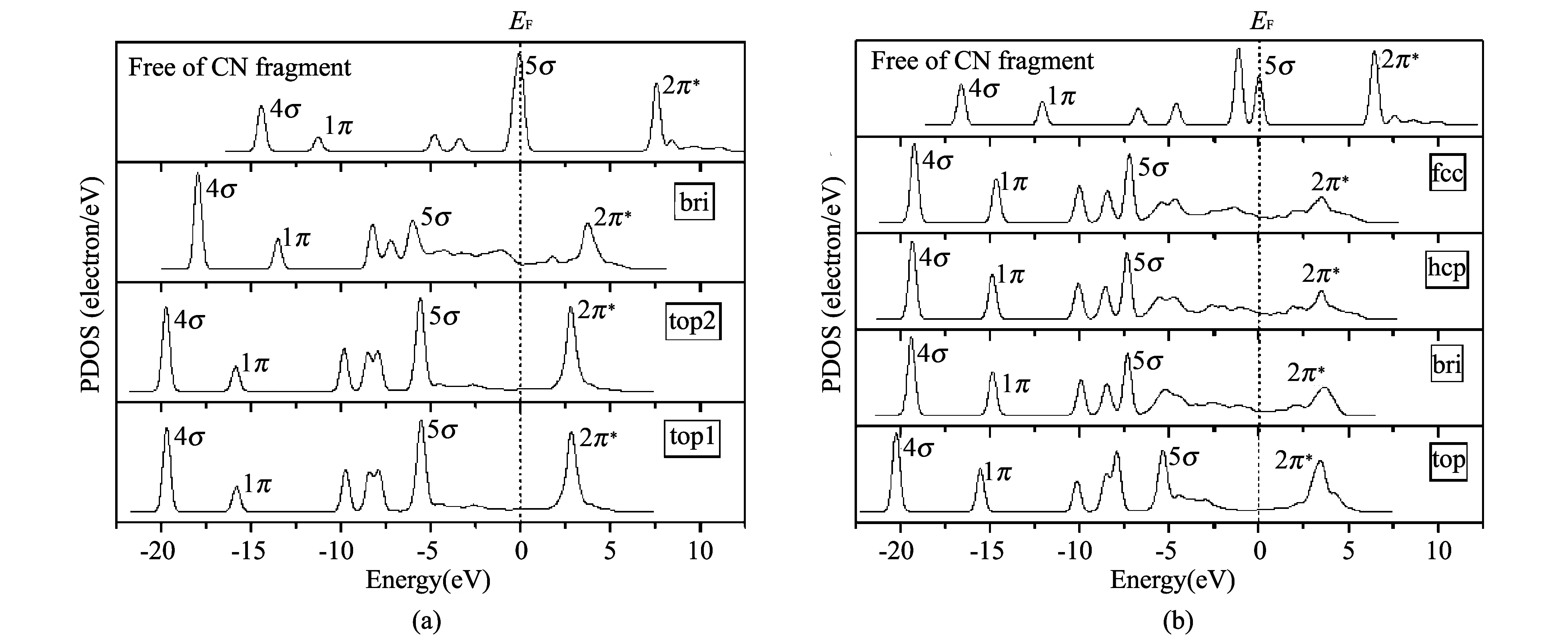

Fig.2 PDOS of the CH3CN (a), CH3NC (b), HCN (c) and HNC (d) adsorption on Pt(111) surface
2.3 HCN and HNC adsorption on Pt(111) surface

3 Electronic structure and density of states

4 Conclusion
The adsorption of CH3CN, CH3NC, HCN and HNC on the Pt(111) surface has been computed at the level of density functional theory for understanding hydrogenation processes of nitriles. At the 1/4 ML low coverage, the most favored adsorption structure for CH3CN is the C—N bond almost parallel to the surface with the C—N bond interaction with adjacent surface Pt atoms. For CH3NC, the most stable configuration is the CH3NC locates at the fcc site with the C-atom bonded to three Pt atoms. For the HCN and HNC adsorption, the adsorption pattern is nearly similarly to the CH3CN and CH3NC, respectively.
The binding mechanism of CO to the Pt(111) surface at the 1/4ML is also analyzed by the DOS analysis. Compared with 5σ and 2π*bands of the free CN fragment, HCN and HNC, the 5σ band of the adsorbed CH3CN, CH3NC, HCN and HNC shifts strongly downwards, and part of the 2π*bands of the adsorbed CH3CN, CH3NC, HCN and HNC lies quite below the Fermi level. This indicates a charge transfer from the Pt(111) surface to the CH3CN, CH3NC, HCN and HNC. As a consequence, C—N bonds are activated.
[2] De Bellefon C, Fouilloux P. Homogeneous and heterogeneous hydrogenation of nitriles in a liquid phase: chemical, mechanistic, and catalytic aspects. Catalysis Reviews: Science and Engineering, 1994, 36(3): 459.
[3] Barrault J, Pouilloux Y. Synthesis of fatty amines. Selectivity control in presence of multifunctional catalysts. Catalysis Today, 1997, 37(2): 137-153.
[4] Huang Y Y , Sachtler W M H. On the mechanism of catalytic hydrogenation of nitriles to amines over supported metal catalysts. Applied Catalysis A:General, 1999, 182(2): 365-378.
[5] Rode C V, Arai M, Shirai M, et al. Gas-phasehyd-rogenation of nitriles by nickel on various supports. Applied Catalysis A: General, 1997, 148(2): 405-413.

[7] Verhaak M J F M, van Dillen A J, Geus J W. The selective hydrogenation of acetonitrile on supported nickel catalysts. Catalysis Letters, 1994, 26(1-2): 37-53.
[8] Medina C F, Tichit D, Coq B, et al. Hydrogenation of acetonitrile on nickel-based catalysts prepared from hydrotalcite-like precursors. Journal of Catalysis, 1997, 167(1): 142-152.
[9] Kishi K, Ikeda S. Adsorption of acetonitrile on evaporated nickel and palladium films studied by X-ray photoelectron spectroscopy. Surface Science, 1981, 107(2/3): 405-416.
[10] Zhuang J, Rusu C N, Yates Jr J T. Adsorption and photooxidation of CH3CN on TiO2. The Journal of Physical Chemistry B, 1999, 103(33): 6957-6967.
[11] Chuang C C, Wu W C, Lee M X, et al. Adsorption and photochemistry of CH3CN and CH3CONH2on powdered TiO2. Physical Chemistry Chemical Physics , 2000, 2(17): 3877-3882.
[12] Avery N R, Matheson T W. Adsorption and decomposition of methyl iso-cyanide on Pt(111). Surface Science, 1984, 143(1): 110-124.
[13] Friend C M, Gavin R M, Muetterties E L, et al. Coordination chemistry of metal surfaces-carbon monoxide chemisorption states on Pt(111). Journal of the American Chemical Society, 1980, 102(5): 1717-1719.
[14] Alexis M, Christian M. Theoretical study of the acetonitrile flip-flop with the electric field orientation: adsorption on a Pt(111) electrode surface. Catalysis Letters, 2003, 91(3-4): 225-234.
[15] Friend C M, Stein J, Muetterties E L. Coordination chemistry of metal surfaces. 2. Chemistry of acetonitrile and methyl isocyanide on nickel surfaces. Journal of the American Chemical Society, 1981, 103(4): 767-772.
[16] Semancik S, Haller G L, Yates J T. The adsorption and dissociation of methyl isocyanide on Rh(111) . Journal of Chemical Physics, 1983, 78(11):6970.
[17] Cavanagh R R, Yates J T. Surface binding of an electronic analog to CO: Infrared evidence for CH3NC chemisorption on Rh/Al2O3. Journal of Chemical Physics, 1981, 75(3): 1551-1559.
[18] Ceyer S T, Yates J T. Orientation of methyl isocyanide adsorbed on silver(311). Journal of Physical Chemistry, 1985, 89(18): 3842-3845.
[19] Payne M C, Allan D C, Arias T A, et al. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Reviews of Modern Physics, 1992, 64(4): 1045-1097.
[20] Milman V, Winkler B, White J A, et al. Electronic structure, properties, and phase stability of inorganic crystals: A pseudopotential plane-wave study. International Journal of Quantum Chemistry, 2000, 77(5): 895-910.
[21] White J A, Bird D M. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations. Physical Review B, 1994, 50(7): 4954-4957.
[22] Perdew J P, Burke S, Ernzerhof M. Generalized gradient approximation made simple. Physical Review Letters, 1996, 77(18): 3865-3868.
[23] Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Physical Review B, 1990, 41(11): 7892-7895.
[24] Monkhorst H J, Pack J D. Special points for brillouin-zone integrations. Physical Review B, 1976, 13(12): 5188-5192.
[25] Hammer B, Hansen L B, N?rskov J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Physical Review B, 1999, 59(11): 7413-7421.
[26] ZHANG Ying-kai, YANG Wei-tao. Comment on Generalized gradient approximation made simple. Physical Review Letters, 1998, 80(4): 890-890.
[27] Gardin D E, Barbieri A, Batteas J D, et al. Tensor LEED analysis of the Ni(111)-p(2×2)-CH3CN structure. Surface Science, 1994, 304(3): 316-324.
[28] Katano S, Herceg E, Trenary M, et al. Single molecule observations of the adsorption sites of methyl isocyanide on Pt(111) by low temperature scanning tunneling microscopy. The Journal of Physical Chemistry B, 2006, 110(41): 20344-20349.
[29] Kang D H, Trenary M. Formation of methylaminocarbyne from methyl Isocyanide on the Pt(111) surface. The Journal of Physical Chemistry B, 2002, 106(22): 5710-5718.
[30] Trenary M, Jentz D, Mills P, et al. The surface chemistry of CN and H on Pt(111) , Surface Science, 1996, 368(1): 354-360.
date: 2012-09-21
Natural Science Foundation of Shanxi Province (No. 2009011014)
REN Jun(junren2003@sxicc.ac.cn)
CLD number: O647 Document code: A
1674-8042(2013)01-0097-06
10.3969/j.issn.1674-8042.2013.01.021
杂志排行

Journal of Measurement Science and Instrumentation的其它文章
- Image enhancement and post-processing for low resolution compressed video
- Randomized Kaczmarz algorithm for CT reconstruction
- Erosion thermocouple temperature acquisition system
- Dynamic calibration method of capacitive pressure measuring device
- Automatic measurement of air-pressure sensor based on two-pressure control instrument
- An experiment to estimate the revolution of DC motor