通过接枝苄基磺酸在MCM-41上固载手性Mn(salen)配合物
2013-10-17胡晨晖江春涛侯文华
李 猛 胡晨晖 江春涛 杨 惠 陈 才 侯文华 陈 静*,
(1南京工业大学理学院,南京 210009)
(2南京大学化学化工学院介观化学重点实验室,南京 210093)
0 引 言
手性环氧化合物是一类用途极广的有机原料和中间体,通过选择性开环或者官能团的转化,可以方便地合成一系列有价值的手性化合物。手性环氧化合物可通过烯烃的不对称环氧化来获得。虽然手性Mn(salen)金属配合物在催化烯烃的不对称环氧化反应中具有活性好、对映选择性高等优点[1-2],但也存在着易分解、难分离、易二聚失活以及不能重复使用等问题。有鉴于此,人们将均相Mn(salen)催化剂负载于各类载体上,并取得了一定的成效[3]。
MCM-41分子筛是一类具有较大的比表面积(>1 000 m2·g-1)、规整有序的孔道结构和较宽孔径(2~10 nm)的多孔材料。利用孔道表面富含的端羟基可对MCM-41进行官能团化改性,从而实现均相催化剂在MCM-41中的组装,达到均相催化剂多相化的目的[4-5]。已有研究表明,将手性催化剂固载于MCM-41分子筛后,在一些催化反应中具有很好的活性及対映选择性,易与产物分离,可重复使用,同时避免了金属配合物的挥发性和毒性[6]。
在MCM-41上固载手性Mn(salen)的方法主要有离子交换法和多步接枝法等[7]。离子交换法需要先对分子筛进行一定的预处理,而且会产生大量的废液;多步接枝法一般则要用到特定的硅烷偶联剂和溶剂,反应条件苛刻,制备过程复杂,成本不菲。此外,所得固载催化剂大多需在昂贵的轴向配体助催化剂参与下才能在不对称环氧化反应中起到满意的催化效果[8-9],限制了它们的实际应用。
本文通过将手性Mn(salen)配合物与苄基磺酸功能化的MCM-41轴向配位,成功获得了一种新型多相手性催化剂;考察了所得催化剂在对α-甲基苯乙烯不对称环氧化反应中的催化性能,发现所得催化剂无需轴向配体助催化剂参与、在温和的条件下就可获得很高的対映选择性。该合成方法所用原料易得,反应条件温和,后处理简单。
1 实验部分
1.1 催化剂的制备
1.1.1 MCM-41 的合成
纯硅MCM-41采用常规水热合成[10]:在室温下,将 2.4 g十六烷基三甲基溴化铵 (CTAB,A.R.级)与12.0 g NaOH 溶液(2 mol·L-1)加入 114 mL 去离子水中搅拌溶解,溶液澄清后,逐滴加入12.2 mL正硅酸乙酯(TEOS,A.R.级)。 1.5 h 后将反应物移入反应釜,于100℃下水热晶化3 d。产物经过滤,去离子水洗涤至中性,70℃下干燥过夜。最后,在540℃焙烧6 h,即得MCM-41分子筛。
1.1.2 苄基磺酸型 MCM-41(BS-MCM-41)的合成[10]
将1 g MCM-41与5 mL苯甲醇、20 mL环己烷加入四口烧瓶,82℃下搅拌回流1 h,过滤,依次用环己烷、丙酮洗涤,60℃干燥过夜,得苄基接枝MCM-41(B-MCM-41)。 用注射器将 0.4 mL ClSO3H 缓缓注入到1 g B-MCM-41和20 mL CHCl3的混合液中,62℃回流1 h,过滤,依次用氯仿、丙酮洗涤,60℃干燥12 h,得苄基磺酸型MCM-41(BS-MCM-41)。反应路线见图式1:1.1.3 手性 Mn(salen)配合物的合成

Scheme 1 Reaction route for preparation of BS-MCM-41
手性Salen配体的合成[11]:在装有搅拌、球形冷凝管和滴液漏斗的100 mL三颈瓶中,加入(1R,2R)-(+)-1,2-环己二胺 L 酒石酸盐(1)(0.74 g,2.8 mmol)、K2CO3(0.78 g,5.6 mmol)和蒸馏水(4 mL),搅拌至溶解后,加入乙醇(15 mL),得浑浊液。加热至回流,将3,5-二叔丁基水杨醛(2)(1.34 g,5.6 mmol, 溶于 15 mL乙醇)慢慢滴入反应体系,滴完后,用乙醇(2 mL)清洗滴液漏斗,继续将黄色浊液回流20 min。停止加热,冷却反应液至室温,再用冰水浴冷却10 min,使晶体析出完全。减压过滤,乙醇洗涤(2 mL),得粗产品2.63 g。粗产品溶解于15 mL二氯甲烷中,用分液漏斗水洗2次(2×8 mL),5 mL 5%盐水洗涤一次。有机相用无水Na2SO4干燥30 min,减压蒸去溶剂,得黄色产品。产品置于60℃烘箱中干燥,得黄色固体(3):Salen 配体(m.p.209.9~211.5 ℃,[α]20D=-322°~-326°)。
Mn(salen)配合物的合成[12]:在配有电磁搅拌器、滴液漏斗和冷凝管的100 mL三口圆底烧瓶中加入40 mL 无水乙醇和 1.0 g(5 mmol)MnCl2·4H2O,加热,搅拌回流,0.5 h 内滴加含 0.9 g(1.67 mmol)Salen配体(3)的15 mL甲苯溶液,继续回流3 h,然后向反应混合物中鼓入大量的空气,溶液变成深褐色,冷却,将其转移到分液漏斗中,加入20 mL甲苯,用3×3 mL去离子水和20 mL饱和食盐水洗涤,无水Na2SO4干燥,减压蒸去溶剂,二氯甲烷-正己烷溶液重结晶,得棕黑色固体(4)即为Mn(salen)配合物。合成路线见图式2。
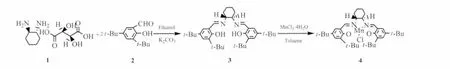
Scheme 2 Synthesis route of chiral Mn(salen)catalyst
1.1.4 手性Mn(salen)配合物修饰的MCM-41的制备[8]
在反应瓶中,加入1.0 g苄基磺酸型分子筛(BSMCM-41)、碳酸氢钠(0.11 g,1.3 mmol)和 20 mL 蒸馏水,室温搅拌3 h。过滤,用蒸馏水洗至中性,得白色粉末(5):苄基磺酸钠修饰的MCM-41(Na-BS-MCM-41)。在反应瓶中加入1.0 g苄基磺酸钠型分子筛,Mn(salen)配合物(4)(1.0 mmol)和乙醇(45 mL),在回流条件下搅拌5 h,过滤,用乙醇和二氯甲烷洗涤,以除去表面吸附的 Mn(salen)催化剂,滤液用 0.1 mol·L-1的AgNO3溶液检测,直到在滤液中检测不到沉淀为止。将固体干燥得棕色产物(6):手性Mn(salen)修饰的MCM-41催化剂(Mn-BS-MCM-41)。反应方程式见图式3。
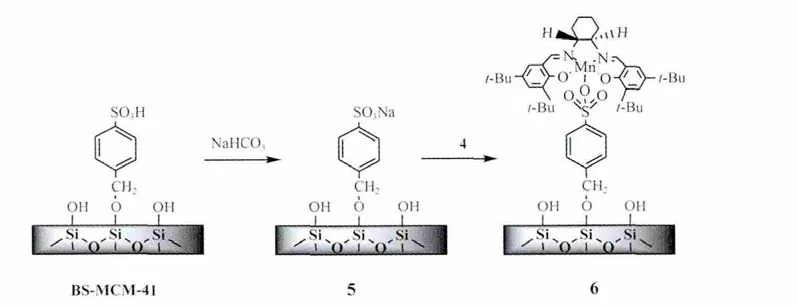
Scheme 3 Preparation of Mn-BS-MCM-41
1.2 催化剂的表征
样品的X射线衍射(XRD)表征在Rigaku公司D/Max-rA型X射线衍射仪上进行,Cu靶,Ni滤波片,Kα 辐射(λ=0.154 18 nm)。 比表面和孔径分布使用Micromeritics ASAP2000型比表面和孔径测定仪测定。红外光谱在Thermo Nicolet公司AVARTE360型红外光谱仪上测定,波数范围为4 000~400 cm-1。锰含量用美国Jarrell-Ash公司J-A1100型电感耦合等离子体发射光谱仪(ICP)测定。热分析采用NETZSCH STA 449 C热分析仪,空气气氛,10℃·min-1的升温速率至800℃。酸量测定采用滴定法[13],即取 0.05 g样品加入到 15 mL 2mol·L-1的食盐水中,连续搅拌并待其平衡后过夜,用0.01 mol·L-1NaOH滴定测其酸量。
1. 3 α-甲基苯乙烯的不对称环氧化反应
将 5 mL 二氯甲烷、1 mmol α-甲基苯乙烯、5 mmol N-甲基吗啉氮氧化物(NMO)和40 μL内标物甲苯加入到25 mL单口烧瓶中,置于冰水浴中搅拌10 min,称取0.02 mmol催化剂倒入烧瓶中,搅拌5 min使催化剂分散均匀。称取2 mmol间氯过氧苯甲酸(m-CPBA)并在30 min内分次加入反应体系,从第一次加入m-CPBA时计时。每1 h取一次样,经处理后直接注入气相色谱仪分析产率和对映选择性。色谱检测条件:手性 β-环糊精柱(RESTEK RTBetaDEXse,30 m×0.25 mm×0.25 μm), 进样器温度230℃,FID检测器,程序升温(初始温度60℃,5℃min-1,终止温度 140 ℃),载气为 N2。
为了考察催化剂的重复使用性能,待反应结束后加入与m-CPBA等物质的量的NaHCO3以中和酸;经旋转蒸发仪旋蒸掉溶剂后,加入去离子水超声溶解旋蒸后的固体,再往水相中加入一定量的正己烷振荡静置后分成两相;先分离出正己烷相,然后水相抽滤、水洗、烘干,得回收催化剂,供下次使用。
2 结果与讨论
2.1 XRD分析
图1为MCM-41和Mn-BS-MCM-41催化剂的XRD 图。 可以看出,两者均在 2θ=2°~3°处出现介孔材料的特征衍射峰,这说明 MCM-41经手性Mn(salen)修饰后,仍然保持了长程有序的六方介孔结构。与MCM-41分子筛相比,手性修饰样品的(100)晶面特征吸收峰向高角度偏移且强度减弱,(110)和(200)晶面特征衍射峰强度明显减弱,这说明 MCM-41经有机基团接枝及手性Mn(salen)修饰后,有序度有所下降。这与手性修饰物进入分子筛孔道内,使孔道结构发生改变及孔径变小等因素有关[14]。
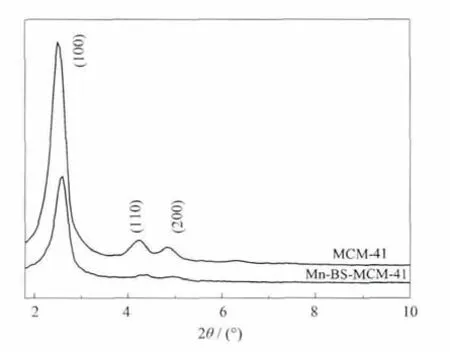
图1MCM-41和Mn-BS-MCM-41的XRD图Fig.1 XRD patterns of MCM-41 and Mn-BS-MCM-41
2.2 低温N2吸附分析
图2 是MCM-41和Mn-BS-MCM-41催化剂的低温N2吸附-脱附等温线和相应的孔径分布曲线。由此测得样品的一些结构参数列于表1。从图2 A可以看出,两个样品的低温N2吸附-脱附曲线均为第Ⅳ类吸附曲线,即典型的介孔材料的吸附-脱附特征曲线, 在 p/p0约为 0.20~0.30之间有一个明显的突跃[15],这是由于N2在介孔中发生毛细管凝聚产生的。MCM-41经苄基磺酸接枝和手性Mn(salen)修饰后,发生突跃的相对压力变小,吸附台阶变得平缓,相关滞后环也发生变化,表明磺酸的引入和手性处理,使孔道的有序性降低,孔径减小,这一结果也与XRD表征结果相符。由孔径分布图(B)可以看出,未修饰的MCM-41具有比较均一的孔道分布,而修饰样品由于晶粒之间的堆积出现了次级孔[10,16]。

图2 MCM-41和Mn-BS-MCM-41的低温N2吸附-脱附等温线(A)和孔径分布曲线(B)Fig.2 N2adsorption-desorption isotherms(A)and pore-size distribution curves(B)of MCM-41 and Mn-BS-MCM-41
从表1可以看出,与MCM-41相比,手性Mn(salen)修饰样品(Mn-BS-MCM-41)的比表面积、孔体积、孔径均减小。这显然与分子筛表面接枝有机基团及手性组分的修饰有关。所得手性修饰Mn-BSMCM-41的比表面积虽较纯MCM-41有所减少,但其比表面积仍可达705 m2·g-1,这为其催化反应物提供了更多的可附着表面。此外,由表1可知Mn-BSMCM-41的平均孔径约1.95 nm,较纯硅样品有所减小。依据晶胞参数a0=23d100/3[17]及平均孔径值,可进一步估算出孔壁厚度δ(δ=a0-Dp),从而得出接枝磺酸后样品的孔壁厚度为2.05 nm,较原纯硅样品的1.62 nm有较明显的增加。
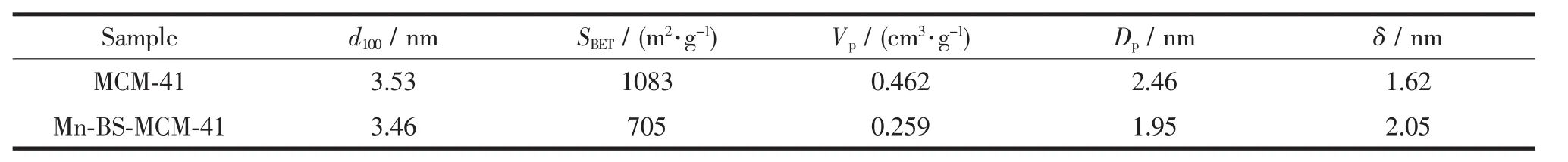
表1 MCM-41和Mn-BS-MCM-41的孔结构性质Table 1 Pore structure properties of MCM-41 and Mn-BS-MCM-41
2.3 FTIR分析
图3为手性Mn(salen)配合物、MCM-41和Mn-BS-MCM-41的红外光谱图。由图可知,手性Mn(salen)配合物在2 954 cm-1附近和1 608 cm-1左右出现了C-H和C=N基团伸缩振动的特征吸收峰,在1 535 cm-1出现Mn(salen)Cl骨架振动峰[8]。MCM-41在807和465 cm-1附近的吸收峰可归属于硅氧四面体Si-O的对称伸缩振动和Si-O的弯曲振动,1 090 cm-1附近的吸收峰归属于外部的Si-O的不对称伸缩振动,而969 cm-1附近窄而尖的吸收峰,归属于表面Si-OH的对称伸缩振动,3 455 cm-1附近为羟基的氢键缔合峰[10,18]。

图3 不同样品的红外光谱图Fig.3 FTIR spectra of different samples
手性修饰后,Mn-BS-MCM-41不但保存了MCM-41的特征峰,而且还出现了接枝基团和手性Mn(salen)配合物的一些特征峰:例如,在3 000 cm-1左右出现了可归属于接枝基团的C-H伸缩振动峰(苯环的烯氢及甲基、亚甲基的饱和C-H伸缩振动峰),1 608、1 535 cm-1附近出现了手性 Mn(salen)配合物的C=N伸缩振动峰和Mn(salen)Cl的骨架特征峰。969 cm-1附近表面Si-OH的对称伸缩振动峰经苄基接枝反应处理后,尖峰基本消失,说明MCM-41表面键合了苄基。另外,一些基团的峰,例如,约为1 240、1 180 cm-1的磺酸SO2不对称伸缩振动和对称伸缩振动特征峰,被MCM-41的Si-O的不对称伸缩振动宽峰覆盖,而不能观察到,但相应的峰形和强度发生了改变。以上结果表明,手性Mn(salen)已经成功接枝负载在MCM-41上。
2.4 热重-差示扫描量热分析
图4为Mn-BS-MCM-41的TG-DSC-DTG图。由图可知,样品Mn-BS-MCM-41在50~100℃间也有一个较小的吸热峰(峰温73℃),并伴随较少的失重,在65℃附近达最大失重速率 (DTG图),失重约1.2%,这同样为样品表面物理吸附水和介孔包裹的水的脱出所致;其在200~600℃间有一温度范围较宽的放热峰(峰温383℃),同时对应TG图上有较多失重,失重约为11.0%。此阶段的失重可归为有机部分的损失[13]。以上结果表明,有机分子已经成功接枝到MCM-41上,且具有一定的热稳定性。

图4 Mn-BS-MCM-41的TG-DSC-DTG图Fig.4 TG-DSC-DTG curves of Mn-BS-MCM-41
通过酸度滴定和ICP测样品的酸量和锰含量,结果表明,MCM-41 的酸量为 0.75 mmol·g-1,主要来自于其表面富含的硅羟基;苄基磺酸接枝后,酸量明显升高到 1.93 mmol·g-1, 手性修饰后, 降至0.57 mmol·g-1, 酸量的变化也证明了手性 Mn(salen)已负载在分子筛上;锰含量为 1.3 mg·g-1,说明手性修饰是成功的。
2.5 催化剂的催化性能
以m-CPBA为氧源,考察了固载催化剂Mn-BSMCM-41和均相Mn(Salen)催化剂在α-甲基苯乙烯的不对称环氧化反应中的催化性能。结果见表2。由表2可知,Mn-BS-MCM-41催化剂在不加轴向配体NMO的情况下,2 h后环氧化物的对映选择性(e.e.值)可高达99%,比均相Mn(Salen)催化剂加NMO得到的 e.e.值 55%高出很多,优于文献报道结果[19,20]。而加入 NMO,其对映选择性(e.e.值)却由原来的 99%下降为12%,转化率也由原来的77%下降到43%。这种反常实验现象文献中也有类似报道[21]。
在烯烃不对称环氧化反应中,大都需要加昂贵的轴向配体助催化剂,才能达到满意的催化效果。轴向配体可以改变活性中间体的几何构型,使其能以最优的构型参与反应,增加底物分子与活性中心的手性识别能力,从而达到提高对映选择性的目的[22]。因此,加入轴向配体NMO的均相Mn(salen)催化剂,可以使α-甲基苯乙烯的不对称环氧化反应的对映选择性(e.e.值)从 18%上升至 55%。
相比于均相Mn(salen)催化剂,固载催化剂Mn-BS-MCM-41中的苄基磺酸基自身就具有很好的轴向配体作用。与NMO相比,苄基磺酸基中的3个氧原子的空间位阻还具撑开立体构型的优势,使氧合金属更加接近于Salen平面(如图5B所示)。当烯烃进攻Salen-Mn(Ⅴ)-O配体时,可以有a(Y)、b(X与Y的中间方向)、c(X)和d(Y)4种方向。其中,途径b取向,即底物烯烃从Schiff碱亚胺处沿着N-Mn键的进攻取向成为首选[23]。N-Mn键轴两侧的电子云密度是不同的,靠芳环一侧电子云密度要比靠环己二胺一侧高。Salen-Mn(Ⅲ)催化效果与烯烃的电子云密度相关,当烯烃沿N-Mn键进攻Mn(Ⅴ)=O时,Salen配体中苯环上的π-电子之间存在一定的排斥力,有利于提高产物的立体选择性。同时,由于底物烯烃α-甲基苯乙烯上带有大空间位阻又属于高π-电子云密度的苯环取代基,从而更有可能得到高的对映选择性[24],最终导致所得Mn-BS-MCM-41催化剂在α-甲基苯乙烯不对称环氧化反应中有高达99%以上的对映选择性以及高效的反应活性。而NMO的加入却使得活性中间体Salen-Mn(Ⅴ)-O周围显得相当拥挤,导致烯烃接近活性中间体Salen-Mn(Ⅴ)-O较为困难,进而影响转化率的提高[21(b)]。至于较低的对映选择性,可能是由于活性中间体Salen-Mn(Ⅴ)-O的最佳构型在载体、连接链、轴向配体NMO和溶剂所构成的微环境中发生扭曲变形,当底物烯烃分子接近活性中间体时,并没有完全按途径b方向进攻,使得活性中间体手性诱导底物时获得R和S构型的机会几乎是一样的,导致手性识别能力和对映选择性下降。

表2 催化剂在烯烃不对称环氧化反应中的催化性能Table 2 Catalytic performance of catalysts for asymmetric epoxidation of olefins
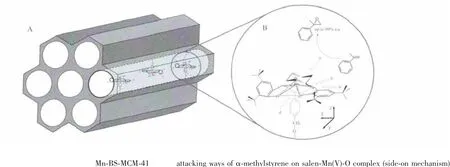
图5 Mn-BS-MCM-41催化剂作用示意图Fig.5 Schematic illustration of the catalytic mechanism on Mn-BS-MCM-41
固载催化剂的重复使用性能是衡量催化活性中心是否有效、稳定地固载于载体上的一个重要依据。为此,我们考察了Mn-BS-MCM-41在α-甲基苯乙烯环氧化反应中的重复使用性能。由表2可以看出,产物的e.e.值随回收次数的增加而相应地减小;催化剂重复使用5次后,产物的对映选择性(e.e.)为70.8%,仍然高于相应的均相催化剂。在回收步骤中,我们采用NaHCO3中和生成的酸,用正己烷萃取出剩余的m-CPBA,通过水洗去生成的盐类化合物。此法较好地保留了活性组分,避免了固载催化剂催化活性的快速衰退,较以往MCM-41负载Mn(salen)配合物的同类催化剂,重复使用性能有了很大的提高[9]。
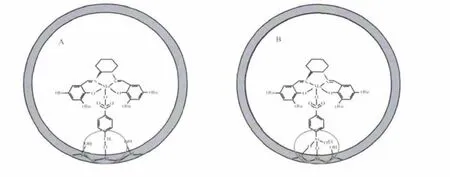
图6 手性Mn(salen)配合物在MCM-41表面的2种固载方式(A-本文,B-文献[19])Fig.6 Schematic illustration of two structure of chrial Mn(salen)complex immobilized on MCM-41 A-this work,B-reference[19]
李灿课题组曾采用多步接枝法,巧妙地通过接枝的烷氧基硅烷上的酚氧、苯磺酸基和丙基磺酸基轴向固载手性Mn(salen)配合物,成功制备了一系列不同结构和良好性能的多相手性不对称环氧化催化剂,探讨了孔径大小、轴的长度、轴的刚柔性等对催化性能的影响,阐明了相应的活性机制,取得了许多有价值的结果[7,19]。与之相比(参见图6),本文的固载方法由于没有使用特定的硅烷偶联剂和溶剂,反应不需要在苛刻的无水条件下进行,后接枝成本经济;由于三乙氧基会占用1个、2个或者3个硅羟基,故苄醇较硅烷偶联剂接枝率更高,有利于手性Mn(salen)的更多固载,从而提高催化性能。另外,接枝的三乙氧基还可能导致分子筛孔道基团繁多杂乱,不利于反应物和产物分子的进出,导致反应效率降低,e.e.值下降[19]。
4 结 论
(1)通过后合成法,利用MCM-41的硅羟基先接枝苄基,通过苄基进一步磺化得到了苄基磺酸型MCM-41。然后,以苄基磺酸为过渡产物,用手性Mn(salen)配合物修饰处理,得到了苄基磺酸轴向固载手性 Mn(salen)配合物的 MCM-41(Mn-BS-MCM-41)。与之前的方法相比,本文方法简单易行,避免了苛刻的实验要求,更具可操作性。
(2)以间氯过氧苯甲酸为氧化剂,考察了所得固载型催化剂对α-甲基苯乙烯的不对称环氧化催化性能,结果表明在0℃反应2 h,无需加入轴向配体NMO的条件下e.e.值可达99%以上,转化率为77%。归因于苄基磺酸基起到了很好的轴向配体作用。经适当处理,固载型催化剂在循环使用5次后e.e.值依然有71%。表现出较好的活性和重复使用性能。
[1]Cavallo L,Jacobsen H.Angew.Chem.Int.Ed.,2000,39(3):589-592
[2]McGarrigle E M,Gilheany D G.Chem.Rev.,2005,105(5):1563-1602
[3]LUO Yun-Fei(罗云飞),ZOU Xiao-Chuan(邹晓川),FU Xiang-Kai(傅相锴).Scientia Sinica Chimica(Zhongguo Kexue:Huaxue),2011,41(3):433-450
[4]Krege C T,Lenonowicz M E,Beck J S,et al.Nature,1992,359(6397):710-712
[5]Hoffmann F,Cornelius M,Morell J,et al.Angew.Chem.Int.Ed.,2006,45:3216-3251
[6]Li C.Catal.Rev.:Sci.&Eng.,2004,46(3/4):419-492
[7]Li C,Zhang H D,Jiang D M,et al.Chem.Commun.,2007:547-558
[8]Zhang H D,Zhang Y M,Li C.J.Catal.,2006,238(2):369-381
[9]Zhao Ji-Quan(赵继全),Zhang Ya-Ran(张雅然),Zhang Yue-Cheng(张月成).Chinese J.Catal.(Cuihua Xuebao),2007,28(1):85-90
[10]CHEN Jing(陈静),HAN Mei(韩梅),SUN Rui(孙蕊),WANG Jin-Tang(王锦堂).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2006,22(9):1568-1572
[11]Larrow J F,Jacobsen E N,Gao Y,et al.J.Org.Chem.,1994,59(7):1939-1942
[12]Zhang H D,Li C.Tetrahedron.,2006,62(28):6640-6649
[13]Dufaud V,Davis M E.J.Am.Chem.Soc.,2003,125(31):9403-9413
[14]RUN Ming-Tao(闰明涛),ZHANG Da-Yu(张大余),WU Gang(吴 刚).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2005,21(8):1165-1169
[15]XU Ru-Ren(徐如人),PANG Wen-Qin(庞文琴).Zeolites and Porous Materials Chemistry(分子筛与多孔材料化学).Beijing:Science Press,2004:146
[16]CHANG Yan(常燕),JIN Sheng-Ming(金胜明),GUAN Hao-Yuan(关豪元).et al.J.Cent.South.Univ.:Science and Technology(Zhongnan Daxue Zirankexue Ban),2008,39(2):265-267
[17]Luechinger M,Frunz L,Prins R,et al.Micropor.Mesopor.Mater.,2003,64(1/2/3):203-211
[18]Ng E P,Subari S N M,Marie O.et al.Appl.Catal.A,2013,450:34-41
[19]Zhang H D,Xiang S,Li C.Chem.Commun.,2005:1209-1211
[20]Huang J,Fu X K,Wang G.et al.Dalton Trans.,2012,41:10661-10669
[21](a)Liao S H,List B.Angew.Chem.Int.Ed.,2010,49:628-631(b)Hu C H,Zhang L H,Hou W H.Catal.Commun.2012.28:111-115(c)Huang X M,Fu X K,Jia Z Y.et al.Catal.Sci.Technol.,2013,3:415-424
[22]ZOU Xiao-Chuan(邹晓川),FU Xiang-Kai(傅相锴),LUO Yun-Fei(罗云飞).Acta Chim.Sin.(Huaxue Xuebao),2011,69(4):431-437
[23](a)Hosoya N,Hatayama A,Katusuki T,et al.Synlett.,1993:641-645(b)Hosoya N,Hatayama A,Katusuki T,et al.Tetrahedron,1994,50:4311-4322
[24]HU Yue-Fei(胡跃飞),LIN Guo-Qiang(林国强).Modern Organic Reaction:Vol.1(现代有机反应:氧化反应).Beijing:Chemical Industry Press,2008:132
