黄嘌呤氧化还原酶钼中心与配体的相互作用
2013-10-13王传铭张旭东
王传铭,张旭东,王 栋,谢 昆
(1.红河学院云南省高校农作物优质高效栽培与安全控制实验室 云南蒙自 661100;2.云南大学生命科学学院 昆明 650091)
作为第6族(VIB族,铬族)的过渡金属,钼(Molybdenum,Mo)是第二过渡系中唯一一种对地球上几乎所有生命都不可或缺的元素[1].在哺乳动物体内,钼主要以钼蝶呤(molybdopterin,MPT)的形式存在于黄嘌呤氧化还原酶(xanthine oxidoreductase,XOR)等三类金属酶中,并以其高的可变化合价参与到氧化还原过程之中.
XOR是广泛分布于各种生物体内调控核酸代谢过程的一种重要酶类,主要功能是催化嘌呤代谢的最后两步,即次黄嘌呤氧化为黄嘌呤和黄嘌呤进一步氧化为尿酸.这些生化反应在氮循环和能量代谢过程中具有重要作用,故有关此酶的结构、功能和活性调控研究一直是生物医学界关注的热点.本文试从结构生物学和计算化学的角度对此酶的催化关键位点(钼蝶呤中心)与其相应配体(底物、抑制剂等)之间的相互作用形式方面的研究进展展开综述,以期帮助人们了解其催化过程及酶活抑制的分子机理.
1 钼碟呤中心的结构与功能
截至2012年7月,PDB(Protein Data Bank)中已经收录了35个实验测定的XOR结构(表1).从这些解析的数据可以看出[2,3],XOR催化位点包含三个氧化还原中心(redox centre):一个钼蝶呤辅因子、两个Fe2S2中心和一个核黄素腺嘌呤二核苷酸辅因子(FAD),三个部分几乎呈线性排列(如图1A).其中钼蝶呤中心是XOR催化关键位点,使体内的黄嘌呤成为尿酸,在此过程中钼中心首先失去2个电子(由六价成为四价),经铁硫中心传递,然后通过FAD将电子传给NAD+或氧分子,将其转化成NADH或.
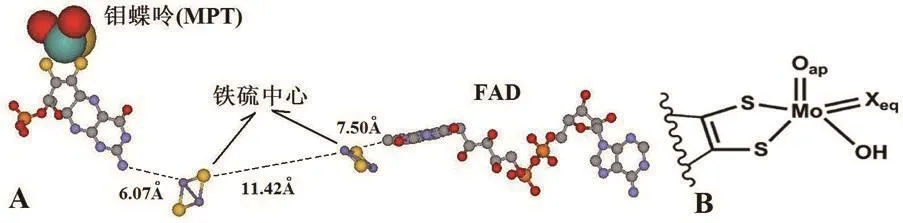
图1 A:XOR的三个辅因子的空间位置关系(以PDB:3UNC为例) B:钼蝶岭辅因子的简化模型
在理论模拟中,钼辅因子中的蝶呤部分常以不同形式简化以减少计算量,其中吡喃环和吡嗪环在模拟中可以提供足够的信息,相比之下蝶呤部分远离钼原子的第二个环可以略去,对计算可靠性影响不大[4].最简化的方式是只保留两个与钼原子配位结合的硫原子及其相连的两个碳原子(图1B).催化关键部位是一个以钼为中心原子的四角锥形配位结构,其具体配位方式人们还存在不同看法理解.相对统一的理解是:四角锥顶端是氧原子(Mo=Oap),底部四边形的三个端点分别是两个与蝶岭亚基结合的硫原子(S-Mo)和一个羟基(Mo-OH).而第四个顶点(Xeq)可能有硫(Mo=Seq)和氧(Mo=Oeq)两种情况.Mo=Seq方式是有活性的结构,也是目前公认的XOR家族的模式特征,被广泛应用在绝大多数理论计算中[5-7].而Mo=Oeq结构没有活性,并且在此种结构中用Sap替换Oap后XOR的活性仍然无法恢复[8,9].但最近也有证据显示,作为一种钼蝶岭的可变结构,Mo=Oeq方式也有其功能[10,11].此外,作为XOR家族的另一成员,乙醛氧化还原酶的钼中心四角锥顶端不是氧原子而是硫原子.

表1 PDB数据库中收录的黄嘌呤氧化还原酶结构信息(截至2012年7月)
2 钼碟呤中心与底物的相互作用
2.1 黄嘌呤生成尿酸的反应循环途径
图2显示了黄嘌呤在钼中心催化下生成尿酸的两种不同的反应机制[12].第一种反应路径依次是图中A-B-C-D-E-F-A,即:黄嘌呤中咪唑环上的氮原子首先与钼原子发生相互作用而靠近(B),随后咪唑环上的碳原子与钼成键(Mo-C方式),该碳原子上的氢原子转移到钼中心的硫基上,Mo-C键和S-H键的形成同步发生(C),属于协同(concerted)反应.来自去质子化水分子或Mo-OH上的羟基进攻底物上这个被激活的碳(D),生成Mo(IV)中间体(E).钼失去一个电子和一个质子(传递给FAD)后形成F.
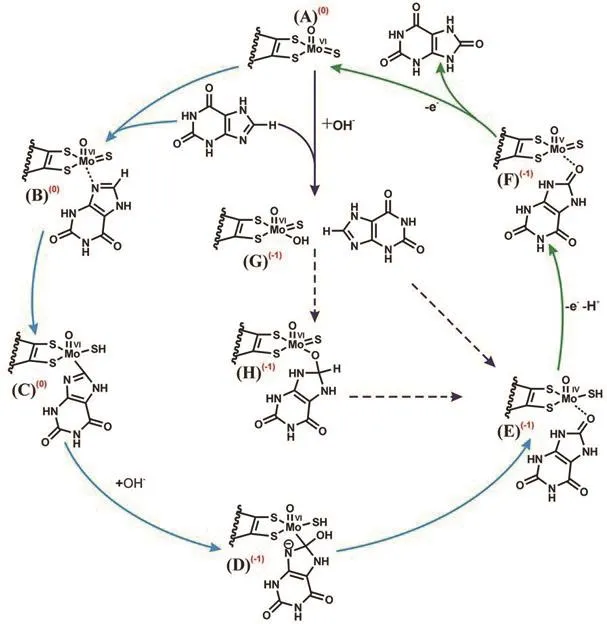
图2 两条不同的黄嘌呤氧化还原酶催化反应循环途径
近年来人们又提出来另一种反应路径(图中的A-G-H-E-F-A),即带有活性羟基的钼中心(G)直接进攻底物上碳原子(Mo-O方式),经过一个中间体(H)或直接一步反应生成E.这两种方式中哪一种更符合真实情况,人们还在不断验证中.
2.2 有关钼中心与底物作用的量化计算和QM/MM研究
最初人们多采用AM1等半经验方法对XOR的反应机制进行量子化学计算[13],密度泛函方法(DFT)应用在此方面始自1997年[14].理论模型中的简化底物有甲醛、乙醛、甲酰胺、黄嘌呤和次黄嘌呤等[15-18].以黄嘌呤为底物的计算只设想了一种协同反应机制[7],以其他较小分子为底物的计算则多给出了协同和分步(stepwise)两种机制(图3).分步方式中第一步反应的势垒(约17.8kcal mol-1)一般高于协同机制(约11.9kcal mol-1)[19].UMP2理论方法计算出的势垒远高于此[20].
氧化还原反应的本质是电子的得失,生物体内的氧化还原酶的主要功能就是通过特定的化学反应在底物分子上添加或移去电子.虽然此功能多是通过酶的辅因子(包括各种辅酶、辅基和金属离子等)参与完成,但组成酶的氨基酸残基在稳定辅因子、识别和稳定配体等方面也具有重要作用.因此,如果把XOR活性位点附近的氨基酸残基的作用也考虑进去,则钼中心催化底物反应的过程更加复杂,相应的理论模拟计算量会急剧增加.因此能够平衡计算效果和计算量的QM/MM方法越来越多地应用在XOR的理论研究中[21,22].钼蝶呤复合物与酶本身并无直接的键键作用,维系二者之间稳定位置关系的主要作用是氢键(如图4).这种以氢键为主的分子间非共价联系也是底物与酶之间十分重要的相互作用形式.图5就列出了黄嘌呤底物在与钼中心反应时可能具有的两种不同的分子取向,分别是美国学者R.Hille提出的“upside”(图5B)方式和日本学者T.Nishino提出的“upside down”(图5A)方式[23].在这两种底物取向中,相互作用的氨基酸残基也不尽相同.精氨酸(Arg880)上带正电的胍基通过静电相互作用稳定了底物在反应中间体中与酶的结合强度,在上述两种取向方式中都具有重要作用.第802位的谷氨酸(Glu802)的作用相对较小,因为实验观察到其突变为谷氨酰胺(Gln802)后,酶活性降低但并未完全消失;而相应的理论计算显示,此时的底物仍以“upside down”取向与突变体结合,

图3 量子化学计算的黄嘌呤氧化还原酶的两种催化反应机制
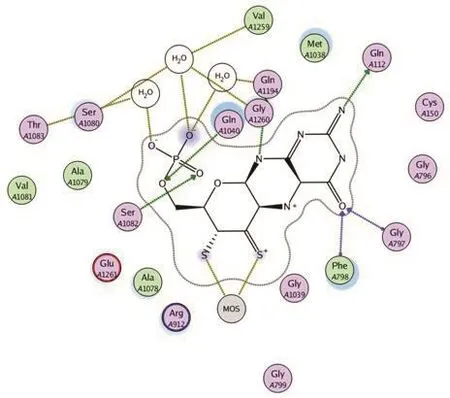
图4 钼蝶呤中心与活性位点附近的氨基酸残基相互作用示意图(以PDB:1FO4为例)
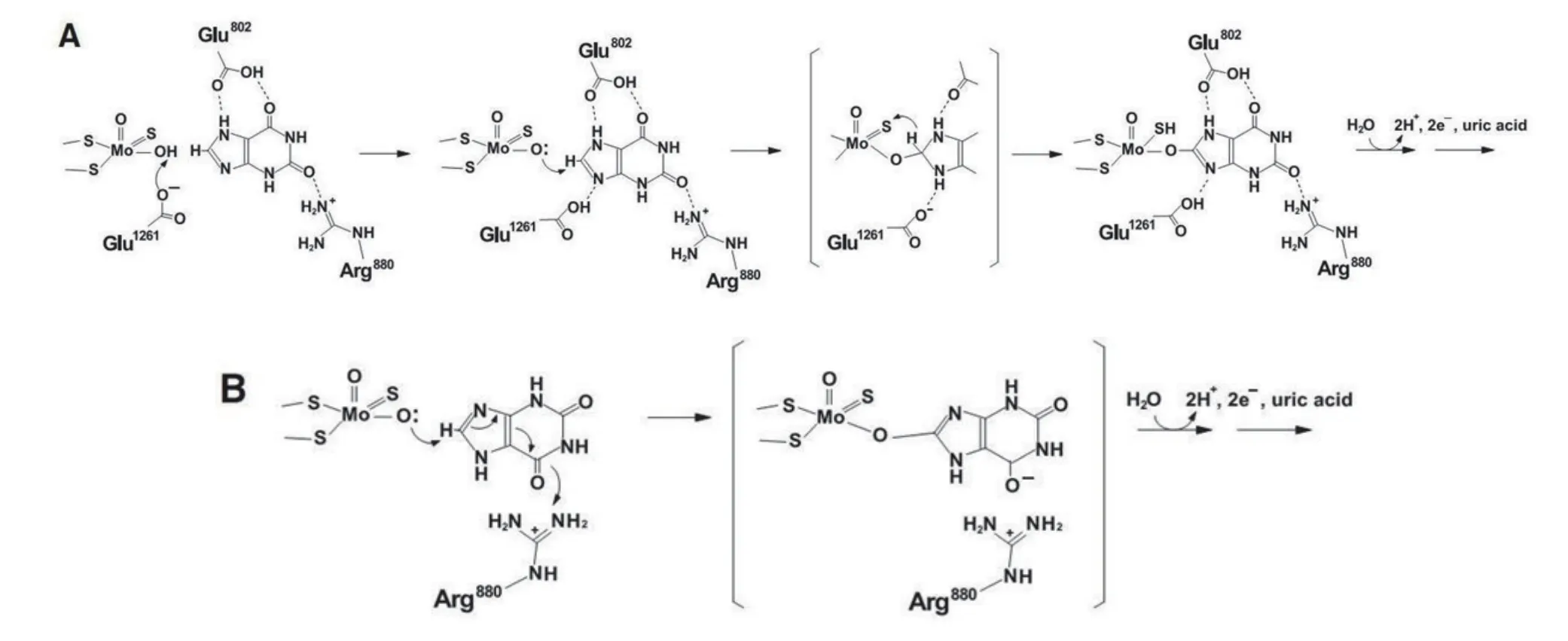
图5 日本学者Yamaguchi等(A)和美国学者Pauff等(B)根据理论计算推测的黄嘌呤底物的两种不同的结合模式(图中氨基酸序号来自大鼠)
只是氢键方式发生了一些改变.2010年T.Nishino又专门通过测定尿酸结合状态下的XOR的晶体结构进一步对这两种黄嘌呤结合取向进行了实验验证[24].
3 钼碟呤中心与抑制剂的相互作用
XOR催化过程中产生的活性氧可参与体内多种生理(如牛乳中富含的XOR使其具有较强的抗菌性[25])和病理(如肿瘤、心血管疾病等)过程.此外,XOR的活性如果异常,会导致体内嘌呤代谢产生的尿酸盐结晶沉积在关节和软组织而引发痛风(gout).由于痛风发病率在以往几十年间都比较低,对XOR抑制剂的研究因而也相对滞后.临床治疗痛风的药物品种极少(截至目前只有2种获批上市),且副作用和不良反应多(如苯溴马隆因潜在的肝毒性已被美国FDA禁止用于痛风治疗).近年来由于人们生活水平逐步提高,痛风的发病率呈逐年上升趋势,成为当今世界中老年常见疾病,被联合国列为21世纪二十大顽症之一,研究开发天然低毒高效的新型XOR抑制剂具有广阔的市场前景[26].
最早发现的XOR抑制剂主要是嘌呤类类似物,包括鸟嘌呤、腺嘌呤、黄嘌呤、次黄嘌呤类似物等,它们与底物结构相似,能竞争性地与酶的结合,从而抑制底物与酶的作用.然而,这些抑制剂与体内其他多种酶的底物也类似,从而会影响到其他酶的功能或经其他酶作用生成毒性代谢产物,限制了该类药物的使用.故1966年以来,临床许可使用的嘌呤类XOR抑制剂仅有别嘌呤醇(allopurinol)一种,虽已上市近半个世纪,至今仍一直在使用.其在体内快速羟化而生成的代谢产物奥西嘌呤(氧化别嘌呤醇,oxypurinol,III期临床阶段)虽抑制活性更强,但其与钼中心的结合是不可逆的Mo-N共价方式(图6A),因而发现新型非嘌呤类、高效、安全的黄嘌呤氧化酶抑制剂显得十分必要而紧迫.
近年来不少学者已经陆续研究发现了一些活性较好的非嘌呤类抑制剂[3,27-30],其中Febuxostat已于2009年由日本武田制药公司生产上市,成为1966年以来FDA批准的第二个抗痛风新药[28].所有这些抑制剂的作用机制各不相同(表2),与底物类似的嘌呤类等化合物对XO产生的多是竞争性(competitive)抑制作用,其他类型化合物对酶的作用则有竞争性、非竞争性(noncompetitive)以及混合型抑制等各种各样不同的类型,此外还存在少量的反竞争性(uncompetitive)抑制类型.竞争性的抑制剂作用位点一般都是底物结合位点(如图6B),酶不能同时与底物和抑制剂结合;非竞争性抑制剂和底物分别在酶的不同结构区域结合,结合没有先后顺序之分.而XOR的反竞争抑制剂只能与酶与底物的复合物发生相互作用,即酶结合上底物之后才能与抑制剂结合,人们对这类抑制剂的确切结合位点还不十分清楚.

图6 两种抑制剂与XOR活性位点之间的作用方式.A:氧化别嘌呤醇 B:FYX-051
在哺乳动物体内,XOR有两种可相互转化的形式即黄嘌呤脱氢酶(xanthenes dehydrogenase,XDH)及黄嘌呤氧化酶(xanthine oxygenase,XO),二者基因序列相同,Fe/S中心和Mo-pt中心的结构也无甚差别,只是FAD周围的氨基酸侧链发生移动,遮盖了NAD+通向FAD的通道(图7),但仍可以允许相对较小的氧分子进入,因此XDH电子传递途径的最终受体是NAD+,而XO电子传递的最终受体是氧分子[31,32].

图7 XOR的两种形式中FAD位点附近的结构差别.A: XDH (PDB:1FO4) B:XO (PDB:1FIQ)

表2 黄嘌呤氧化还原酶抑制剂
有确凿的实验结果证明,XO催化反应类似ping-pong机制[33],即第一种底物黄嘌呤先结合到酶的钼中心位点上,使酶构型瞬间发生改变,并释放出第一种产物尿酸,之后电子受体氧分子才结合到酶的FAD位点上并释放第二种产物双氧水,同时释放出酶,从而开始下一轮催化.氧分子的这种结合方式在表现形式上非常类似反竞争性抑制剂.据此我们推测反竞争性抑制剂与XOR的结合方式:一种可能类似氧分子,结合在变构后的FAD位点附近,阻断电子受体的进入,从而发挥抑制活性;另一种是直接和钼碟呤-黄嘌呤复合体结合,阻止尿酸的生成和释放.我们希望未来能通过一系列的实验验证和构效关系分析去逐步探索这个问题.
[1]Young CG.Molybdenum: MPT-Containing Enzymes[G].//King RB.Encyclopedia of Inorganic and Bioinorganic Chemistry.Wiley,2011:Vol 5.
[2]Hille R, Nishino T, Bittner F.Molybdenum Enzymes in Higher Organisms.Coord Chem Rev.2011,255(9-10):1179-1205.
[3]Okamoto K, Matsumoto K,Hille R,et al.The Crystal Structure of Xanthine Oxidoreductase During Catalysis: Emplications for Reaction Mechanism and Enzyme Inhibition[J].Proc Natl Acad Sci USA.2004,101(21):7931-7936.
[4]Ryde U, Schulzke C, Starke K.Which Functional Groups of the Molybdopterin Ligand Should be Considered When Modeling the Active Sites of the Molybdenum and Tungsten Cofactors? A Density Functional Theory Study[J].J Biol Inorg Chem.2009,14(7):1053-1064.
[5]Doonan CJ, Stockert A, Hille R,et al.Nature of the Catalytically Labile Oxygen in The Active Site of Xanthine Oxidase[J].J Am Chem Soc.2005,127(12):4518-4522.
[6]Schwarz G, Mendel RR.Molybdenum Cofactor Biosynthesis and Molybdenum Enzymes[J].Annu Rev Plant Biol.2006;57:623-47.
[7]Bayse CA.Density-Functional Ttheory Models of Xanthine Oxidoreductase Activity:Comparison of Substrate Tautomerization and Protonation [J].Dalton Transactions.2009,0(13):2306-2314.
[8]Huber R,Hof P,Duarte RO,Moura JJ, et al.A Structure-Based Catalytic Mechanism for the Xanthine Oxidase Family of Molybdenum Enzymes[J].Proc Natl Acad Sci USA.1996,93(17):8846—8851.
[9]Massey V,Edmondson D.On the Mechanism of Inactivation of Xanthine Oxidase by Cyanide[J].J Biol Chem.1970,245(24):6595—6598.
[10]Santos-Silva T,Ferroni F,Thapper A,et al.Kinetic,Dtructural,and EPR Dtudies Reveal That aldehyde Oxidoreductase From Desulfovibrio Gigas Does not Need A Sulfido Ligand for Catalysis and Give Evidence for a Direct Mo-C Interaction in a biological System[J].J Am Chem Soc.2009,131 (23):7990-7998.
[11]Ilich P and Hille R.Oxo, Sulfido, and Tellurido Mo-enedithiolate Models for Xanthine Oxidase:Understanding the Basis of Enzyme Reactivity[J].J Am Chem Soc.2002,124 (24):6796-6797.
[12]Metz S and Thiel W.Theoretical Studies on the Reactivity of Molybdenum Enzymes[J].Coord Chem Rev.2011,255( 9-10):1085-1103.
[13]Rastelli G,Costantino L,Albasini A.A Model of the Interaction of Substrates and Inhibitors with Xanthine Oxidase[J].J Am Chem Soc.1997,119 (13):3007-3016.
[14]Bray MR, Deeth RJ.The Catalytic Activity of Xanthine Oxidase:Mechanistic Insights Through Computer Modelling[J].J Chem Soc,Dalton Trans.1997, (8):1267-1268.
[15]Voityuk AA, Albert K, Romao MJ, et al.Substrate Oxidation in the Active Site of Xanthine Oxidase and Related Enzymes.A Model Density Functional Study[J].Inorg Chem.1998, 37 (2):176-180.
[16]Amano T, Ochi N, Sato H,et al.Oxidation Reaction by Xanthine Oxidase.Theoretical Study of Reaction Mechanism[J].J Am Chem Soc.2007 129 (26), 8131-8138.
[17]Shanmugam M.Zhang B, McNaughton RL,et al.The Structure of Formaldehyde-Inhibited Xanthine Oxidase Determined by 35 GHz 2H ENDOR Spectroscopy[J].J Am Chem Soc.2010,132 (40):14015-14017.
[18]Hemann C,Ilich P, Stockert AL, et al.Resonance Raman Studies of Xanthine Oxidase: the Reduced Enzyme—Product Complex with Violapterin[J].J Phys Chem B.2005,109 (7):3023-3031.
[19]Zhang XH, Wu YD.A Theoretical Study on the Mechanism of the Reductive Half-Reaction of Xanthine Oxidase[J].Inorg Chem.2005,44 (5):1466-1471.
[20]Ilich P,Hille R.Mechanism of Formamide Hydroxylation Catalyzed bya molybdenum-Dithiolene Complex:A Model for Xanthine Oxidase Reactivity[J].J Phys Chem B.1999,103(25):5406-5412.
[21]Metz S, Thiel W.A Combined QM/MM Study on the Reductive Half-Reaction of Xanthine Oxidase: Substrate Orientation and Mechanism[J].J Am Chem Soc.2009,131 (41):14885-14902.
[22]Metz S, Thiel W.QM/MM Studies of Xanthine Oxidase: Variations of Cofactor,Substrate,and Active-Site Glu802[J].J Phys Chem B.2010,114 (3):1506-1517.
[23]Nishino T,Okamoto K,Eger BT,etal.Mammalian Xanthine Oxidoreductase—Mechanism of Transition From Xanthine Dehydrogenase to Xanthine Oxidase[J].FEBS J.2008,275(13):3278-3289.
[24]Okamoto K,Kawaguchi Y,Eger BT,et al.Crystal Structures of Urate Bound Form of Xanthine Oxidoreductase: Substrate Orientation and Structure of the Key Reaction Intermediate[J].J Am Chem Soc.2010,132 (48):17080-17083.
[25]Massey V,Brumby PE,Komai H.Studies on Milk Xanthine Oxidase.Some Spectral and Kinetic Properties[J].J Biol Chem.1969,244(7):1682~1691.
[26]Pacher P,Nivorozhkin A, Szabó C.Therapeutic Effects of Xanthine Oxidase Inhibitors: Renaissance Half A Century After the Discovery of Allopurinol[J].Pharmacol Rev.2006,58(1):87-114.
[27]Borges F, Fernandes E, Roleira F.Progress Towards the Discovery of Xanthine Oxidase Inhibitors[J].Curr Med Chem.2002,9(2):195-217.
[28]苏彦雷,蒋建勤,汪俊松.等.黄嘌呤氧化还原酶抑制剂的构效关系研究进展[J].药学进展,2009,33(8):350-359.
[29]Noro T,Ueno A, Mizutani M,etal.Inhibitors of Xanthine Oxidase From Athyrium Mesosorum[J].Chem Pharm Bull (Tokyo).1984,32(11):4455-4459.
[30]Hu LN, Hu HG,WuWF,et al.Discovery of Novel Xanthone Derivatives As Xanthine Oxidase Inhibitors[J].Bioorg Med Chem Lett.2011,21(13):4013-4015.
[31]Kuwabara Y,Nishino T,Okamoto K,etal.Unique Amino Acids Ccluster For Switching From the Dehydrogenase to Oxidase Form of Xanthine Oxidoreductase[J].Proc Natl Acad Sci USA.2003,100 (14):8170-8175.
[32]McManaman JL, Bain DL.Structural and Conformational Analysis of the Oxidase to Dehydrogenase Conversion of Xanthine Oxidoreductase[J].J Biol Chem.2002,277(24):21261-21268.
[33]Harris CM,Massey V.The Reaction of Reduced Xanthine Dehydrogenase with Molecular Oxygen.Reaction kinetics and Measurement of Superoxide Radical.J Biol Chem.1997,272(13):8370-8379.
