拟反相液相色谱法快速测定奶茶中咖啡因含量的研究
2013-10-08董丽丽王力川
董丽丽,于 玲,李 敏,王力川
(邢台学院 生物化学系,河北 邢台 054001)
奶茶兼具牛奶和茶的双重营养,由于美味可口、时尚休闲、方便快捷,深受广大消费者的喜爱,尤其是以青少年消费群体为主。近年来,国内奶茶市场正处在快速发展阶段,多种品牌及不同口味的奶茶层出不穷。奶茶中含有生物碱、多种维生素、单宁酸、芳香油等300多种对人体有益的化学成份。咖啡因是奶茶中的重要生物碱,具有解除疲劳、兴奋神经、促进血液循环等作用,但大剂量或长期使用会损害人体健康,有成瘾性,被列入受国家管制的精神药品范围。因此对食品中咖啡因的含量检测是非常必要的。在咖啡因检测中,最常见的是咖啡、茶、软饮料中咖啡因的测定[1-10],对奶茶中咖啡因的检测则相对较少。拟反相色谱法[11]是以正相色谱所用的极性色谱柱为固定相,配以反相色谱的含水溶剂为流动相的色谱方法。相比于反相色谱和亲水作用色谱,拟反相色谱的研究极少,已有报道主要是对保留行为的探讨[12-16],而利用拟反相色谱进行检测的文献并不多。本实验将拟反相液相色谱法用于奶茶中咖啡因含量的测定,建立的方法所需有机相含量极低(仅占5%),且分析速度快(5 min内可完成样品的分离测定),极大地减少了有机试剂的耗费,同时也减少了对环境的污染。本研究是对食品中咖啡因检测方法的补充,可用于样品的日常测定,同时也可以促进色谱机理的进一步探讨。
1 实验部分
1.1 仪器条件与试剂
HPLC条件:岛津LC-20AT高效液相色谱仪配二极管阵列检测器;色谱柱为InertsilHPLC Col-umn 100-5SIL(150mm×4.6mmi.d.5μm);流动相为0.03mol·L-1甲酸钠溶液(pH 2.9)-乙腈(95∶5),流速1.0mL·min-1,进样量10μL。检测波长274 nm。
咖啡因标准品由茶叶提取,升华提纯。取适量溶于甲醇制备成5.133 g·L-1的标准储备液,使用时用流动相稀释至所需浓度。
12种不同品牌、不同口味及包装(袋装或杯装)的奶茶均购自当地一家大型超市。
乙腈、甲醇、异丙醇均为色谱纯,其他试剂为分析纯,实验用水为超纯水。
1.2 样品的制备
准确称取各种奶茶约1 g,用适量85℃的水溶解后,用水定容至100 mL。移取10 mL离心,取上清液经0.45 μm滤膜过滤后,吸取10 μL进样测定。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 有机溶剂类型的选择 本文采用拟反相色谱法测定咖啡因。首先考察了流动相中有机溶剂类型对咖啡因保留的影响。保持其它条件相同,分别以甲醇、异丙醇、乙腈3种有机溶剂与甲酸钠缓冲液(0.03 mol·L-1,pH 2.9)(5 ∶95)组成流动相,所得色谱图见图1。3种有机溶剂对溶质的洗脱强度按以下顺序递增:甲醇<异丙醇<乙腈。

图1 有机溶剂类型对咖啡因保留的影响Fig.1 Influence of organic modifier on the retention of caffeine A.acetonitrile;B.isopropanol;C.methanol
由于甲醇是质子化溶剂,具有强的氢键作用能力,在硅胶表面可与水竞争吸附,流动相中的甲醇一部分吸附在固定相表面代替水分子。该条件下,流动相中甲醇的含量很低(只占5%),此时会更低。而溶质的保留对流动相中有机相的含量极为敏感[12]。因此甲醇含量的轻微变化可导致溶质保留的迅速改变。3种溶剂中甲醇的氢键作用倾向大,吸附到固定相上的数量多,相当于流动相中有机相的含量降低,所以咖啡因的保留增强。与甲醇相比,异丙醇由于碳链的增长使其亲水性降低,与固定相表面活性点的作用减弱,因而遗留在流动相中的数量相比甲醇要多,洗脱能力增强,所以咖啡因在其中的保留比在甲醇中弱。而乙腈是非质子化溶剂,不与固定相表面的硅羟基作用,存在于流动相中的乙腈数量相对较多,相当于流动相洗脱能力增强,所以咖啡因在其中的保留时间最短。
实验结果表明,有机溶剂的类型对咖啡因的保留有着重要影响。由于待测物在乙腈中的洗脱时间最短,所得色谱峰最为尖锐、柱效高(n=31 600/m)、峰形对称性好(As=1.161),而在其它两种体系中均出现严重拖尾现象,因此选择乙腈体系测定咖啡因。
2.1.2 有机溶剂含量的选择 进一步考察了流动相中乙腈含量对咖啡因保留值的影响(见图2)。在乙腈含量小于50%时,随流动相中乙腈含量的增加,溶质的保留值下降,表现出类似反相色谱的特性。类似结果也出现在阿替洛尔的分析中[17],因此选择乙腈的最佳含量为5%。
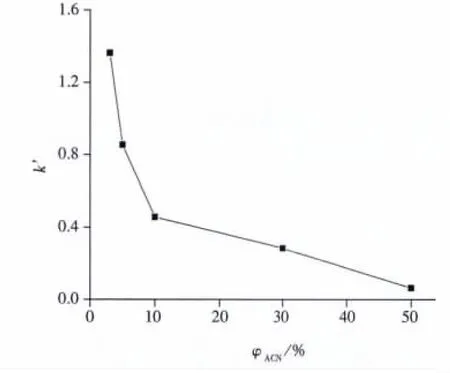
图2 流动相中乙腈含量对咖啡因保留因子的影响Fig.2 Effect of ACN content in mobile phase on retention factors of caffeine
2.1.3 缓冲溶液pH值的选择 流动相pH值同时影响到溶质和硅胶表面硅羟基的电离,进而影响溶质的保留。本实验考察了pH值对咖啡因保留因子的影响。当pH值由2.5升至6.4的过程中,咖啡因的洗脱时间先随pH值的增加而延长,达到最大值后,随pH值的增加,保留反而减弱(见图3),说明拟反相色谱中溶质的保留机理是混合机理,除了分配作用,还存在一定的离子交换作用[16]。

图3 pH值对咖啡因保留因子的影响Fig.3 Effect of pH value of mobile phase on retention factors of caffeine
2.1.4 离子强度的影响 由图4可以看出,随体系中甲酸钠浓度的增加,咖啡因的保留时间不断缩短。说明离子强度增加时,体系中带正电荷的阳离子可与质子化的咖啡因竞争固定相上的活性保留中心,导致溶质的保留值减弱。与上述由pH值体现的拟反相色谱中溶质的保留机理是基于分配作用和离子交换作用的双重作用相一致。
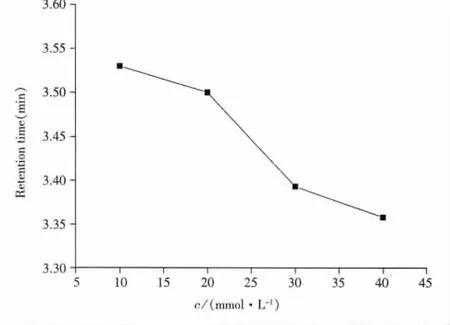
图4 体系中离子强度对咖啡因保留时间的影响Fig.4 Effect of ionic strength in mobile phase on retention time of caffeine
通过上述实验,结合保留强度和柱效等因素确定测定咖啡因的流动相为:乙腈与甲酸钠缓冲液(0.03 mol·L-1,pH 2.9)(5 ∶95)。
2.2 线性范围、检出限、重现性及回收率
将配制好的系列浓度咖啡因标准溶液,按上述色谱条件进样,以峰面积对咖啡因浓度(mg·L-1)作线性回归分析。结果显示,咖啡因的线性范围为0.2~1 000 mg·L-1,相关系数(r)为0.999 5,检出限(S/N=3)为0.05 mg·L-1。我国目前的国标中尚无对食品中咖啡因含量的规定(只有一个对于可乐饮料的咖啡因限定,含量不超过0.15 g·kg-1)[18]。该方法较宽的线性范围可以满足不同食品中咖啡因含量的测定。
精密吸取20 mg·L-1咖啡因标准溶液10 μL,进样10次平行测定,测得峰面积的相对标准偏差为1.8%,保留时间的相对标准偏差为1.6%,说明该方法的重现性好。
在已知咖啡因含量的奶茶制备液中加入咖啡因标准液适量,加水稀释、定容,过滤后吸取10 μL进样,记录色谱图,每个样品平行进样5次,计算方法的回收率与相对标准偏差(见表1)。由表1可知,平均回收率为98%~103%,相对标准偏差为1.9%~3.6%,说明本方法具有很好的准确度和精密度。

表1 奶茶中咖啡因的加标回收率与相对标准偏差(n=5)Table 1 Recoveries and relative standard deviations(RSDs)of caffeine in milk tea(n=5)
2.3 实际样品的测定
奶茶中的茶粉含有咖啡因,将所建立的拟反相液相色谱法用于12种奶茶中咖啡因含量的测定,以检验方法的实用性。所测12个样品均经溶解、离心、过滤处理后进样测定,所得谱图见图5。由图5可见,尽管不同品牌的奶茶组成复杂,成分各异,但除了目标物咖啡因的保留较强外,大多数组分在该色谱模式下几乎无保留或保留很弱,使得咖啡因均可与共存干扰组分实现基线分离,表明该方法对咖啡因测定具有高选择性。表2中列出了12种奶茶中咖啡因的含量。结果显示,12种奶茶中咖啡因的含量在0.386 1~1.617 2 g·kg-1之间。目前,对于奶茶中咖啡因含量测定的研究未见报道,尚无数据可比较。国家卫生标准可乐饮料的咖啡因限定含量为不超过0.15 g·kg-1[18]。如果将奶茶看作饮料,则其中咖啡因的含量均超出标准值;若当作茶看待,则低于茶叶中咖啡因的含量(一般<40 g·kg-1)。但喜欢喝奶茶的人群主要为儿童、青少年,所以奶茶中的咖啡因需要谨慎对待。

图5 奶茶中咖啡因的色谱图Fig.5 Chromatograms of caffeine in milk tea the numbers denoted were the same as those in Table 2

表2 市售奶茶中咖啡因的测定结果(n=5)Table 2 Analytical result of caffeine in commercially available samples(n=5)
3 结论
本文利用拟反相液相色谱法快速测定了奶茶中咖啡因的含量,系统地探讨了影响咖啡因保留的因素。与反相液相色谱法相比,硅胶色谱柱更为价廉,且该方法的分析速度快,在流动相中只需极少量的有机溶剂,即可实现快速高通量连续运行,极大节约了测试成本,同时降低了对环境的污染。该法的线性范围宽、灵敏度好、回收率高、精密度好,可用于多种奶茶中咖啡因的测定。
[1] vorc L,Tomcˇík P,Svítková J,Rievaj M,Bustin D.Food Chem.,2012,135(3):1198 -1204.
[2] Guo S J,Zhu Q Q,Yang B C,Wang J,Ye B X.Food Chem.,2011,129(3):1311 -1314.
[3] Alpdogan G,Karabina K,Sungurd S.Turk J.Chem.,2002,26(2):295-302.
[4] Najafi N M,Hamid A S,Afshin R K.Microchem.J.,2003,75(3):151 -158.
[5] Chen C N,Liang C M,Lai J R,Tsai Y J,Tsay J S,Lin J K.J.Agric.Food Chem.,2003,51(25):7495 -7503.
[6] Srdjenovic B,Djordjevic-Milic V,Grujic N,Injac R,Lepojevic Z.J.Chromatogr.Sci.,2008,46(2):137 -143.
[7] Zuo Y G,Chen H,Deng Y W.Talanta,2002,57(2):307-316.
[8] Hideki H,Atsushi N,Tomomi U,Katsunori K.J.Chromatogr.A,2002,942(1/2):271-273.
[9] Pura Naik J.J.Agric.Food Chem.,2001,49(8):3579 -3583.
[10] Wang J Q,Jia L,Xia M.Mod.Food Sci.Technol.(王嘉琦,贾丽,夏敏.现代食品科技),2011,27(1):114-116.
[11] Crommen J.J.Chromatogr.,1979,186:705 -724.
[12] Cox G B,Stout R W.J.Chromatogr.A,1987,384(2):315-336.
[13] Gritti F,Pereira A S,Sandra P,Guiochon G.J.Chromatogr.A,2009,1216(48):8496-8504.
[14] Gritti F,Pereira A S,Sandra P,Guiochon G.J.Chromatogr.A,2010,1217(5):683-688.
[15] Li Y Y,Li J,Chen T,Liu X Y,Zhang H X.J.Chromatogr.A,2011,1218(11):1503-1508.
[16] Li Y Y,Li J,Chen T,Liu X Y,Zhang H X.Anal.Chim.Acta,2012,726:102-108.
[17] Dong L L,Wang C P.Chin.J.Anal.Lab.(董丽丽,王春平.分析试验室),2013,32(1):114-116.
[18] GB 2760 -2011.Hygienic Standards for Uses of Food Additives.National Standards of the Peoples Republic of China(食品添加剂使用卫生标准.中华人民共和国国家标准).
