聚苯胺改性氮掺杂碳纳米管制备及其超级电容器性能
2013-09-21李莉香安百钢
李莉香 陶 晶 耿 新 安百钢
(辽宁科技大学化工学院,材料电化学研究所,辽宁鞍山114051)
1 引言
碳纳米管由于其独特的中空结构、高的比表面积、良好的导电性、优异的力学、电化学和场发射性能1-6等,使其在诸多领域有着良好的应用前景.近年来,通过对碳纳米管进行氮掺杂,以提高碳纳米管的应用性能,成为碳纳米管合成和应用领域的研究热点之一.对碳纳米管进行氮掺杂不仅可以改善其水分散性,7-9从而解决碳纳米管在实际应用中难分散的缺点,而且氮掺杂原子能够改变碳纳米管局部电荷密度,提高碳纳米管的电子传递性,降低电阻系数.10在碳纳米管作为超级电容器电极的应用领域,由于氮掺杂引入的含氮官能团能够带来准法拉第效应,可明显提高碳纳米管超级电容器的比容量.11-15在燃料电池催化剂载体研究领域,最近科学家们发现碳纳米管中的氮掺杂原子本身还具有氧还原催化活性,16-18从而使非贵金属负载的氮掺杂碳纳米管作为燃料电池电极的研发也备受关注.
目前,对碳纳米管进行氮掺杂主要有两种方式,一种是在合成碳纳米管过程中,通过使用含氮前驱体作为碳源实现对碳纳米管的氮掺杂,但热解过程中大量氮以气体的形式损失而难以获得高的氮掺杂量,并且去除催化剂的过程通常会破坏碳纳米管本征结构;19,20另一种是通过对合成的碳纳米管表面化学改性进行氮掺杂,如强酸或NH3等化学处理方法21-23可实现对碳纳米管的有效氮掺杂,但此种处理方式在引入含氮官能团的同时,也会使碳纳米管的原有结构遭到破坏,导致碳纳米管导电性降低.24,25采用含氮有机溶剂对碳纳米管进行氮掺杂是一种相对温和的方法,由于碳纳米管与有机溶剂具有很好的相溶性,有利于对碳纳米管进行均匀氮掺杂,并且不会对碳纳米管产生破坏作用,从而可实现在氮掺杂的同时,保持碳纳米管的原有结构和优异的力学特征,但其掺氮量较低.26
本文采用聚苯胺改性处理制备氮掺杂碳纳米管,即通过苯胺原位化学聚合在碳纳米管表面形成聚苯胺包覆层,然后通过炭化处理使聚苯胺转化形成氮掺杂碳层,从而制备出氮掺杂碳纳米管,而碳纳米管的本征结构未遭任何破坏,并通过改变苯胺用量,制备出不同氮掺杂层厚度的碳纳米管.
2 实验方法
多壁碳纳米管由日本三好有限会社提供.经检测不含金属铁、镍等催化剂及其它如噻吩之类的生长促进剂.未进行聚苯胺改性处理的原始碳纳米管记为CNT.本文中所使用的试剂均为分析纯,国药集团化学试剂有限公司生产.制备氮掺杂碳纳米管的具体实验方法如下:将0.30 g苯胺单体溶于47.3 mL浓度为1 mol·L-1的盐酸溶液中,磁力搅拌使苯胺分散均匀,之后将0.70 g碳纳米管加入到上述溶液中,超声处理0.5 h获得碳纳米管悬浮液.另外,将0.70 g过硫酸铵溶于36.6 mL浓度为1 mol·L-1的盐酸溶液中,将该溶液于冰水混合浴中冷却至0-5°C.将上述步骤制备的碳纳米管悬浮液置于冰水混合浴中搅拌,同时缓慢滴加过硫酸铵溶液.产物经真空过滤,用去离子水清洗过滤,然后在86°C下真空干燥24 h,得到聚苯胺/碳纳米管复合物.为了考察苯胺用量对碳管表面形成的氮掺杂层厚度和超级电容器的比电容量等的影响,将苯胺单体用量和盐酸溶液用量同时减半或加倍,其它合成条件不变,即制备苯胺用量分别为0.15和0.60 g的另外两个聚苯胺/碳纳米管复合物样品.依据苯胺用量从低到高(苯胺量分别为0.15,0.30和0.60 g)的样品分别记为PANI/CNT-1、PANI/CNT-2和 PANI/CNT-3.将制备的聚苯胺/碳纳米管复合物置于管式热处理炉中,在氮气气氛下以4°C·min-1从室温升温到400°C并恒温2 h,以去除样品中水分,同时稳定碳管表面包覆层,再以4 °C·min-1升到700 °C并恒温1 h,以除去氢、氧等非碳原子,使碳管表面包覆层炭化,然后自然冷却到室温,取出样品,经上述处理得到的碳纳米管样品按苯胺用量从低到高记为NCNT-1、NCNT-2和NCNT-3.
原始碳纳米管、聚苯胺包覆碳纳米管及氮掺杂碳纳米管形貌观察采用JEOL 2010透射电镜.分别用ESCALAB250表面分析系统(铜靶,X射线的能量为284.5 eV;结合能扫描范围为0-1350 eV)的X射线光电子谱(XPS),显微激光拉曼光谱仪(JY Labram HR 800)和ASAP2010M比表面和孔结构分析仪分析聚苯胺改性氮掺杂处理前后碳纳米管的结构特征、表面化学组成、氮掺杂量和比表面积.
聚苯胺改性氮掺杂处理前后的碳纳米管作为超级电容器电极材料的电化学性能测试采用三电极测试体系.其中,Hg/HgO电极作为参比电极,表观面积为6 cm2的铂网为辅助电极,分别以原始碳纳米管和氮掺杂碳纳米管为电极活性物质制备无导电添加剂的研究电极,其制备方法为:将原始碳纳米管或氮掺杂碳纳米管与聚四氟乙烯(PTFE)粘结剂按质量比9:1均匀混合,之后将其平铺于两片泡沫镍之间,中间放一条镍带作为导线引出,然后在2 MPa压力下压制成电极片(10 mm×10 mm),作为研究电极备用.电化学测试前,将研究电极浸泡在6 mol·L-1的KOH电解液中真空抽滤以抽去其中夹杂的气泡,并浸泡24 h.电化学测试采用CHI660A电化学分析系统完成.
3 结果与讨论
3.1 氮掺杂处理前后碳纳米管的形貌
原始碳纳米管和聚苯胺包覆碳纳米管炭化前后样品的形貌如图1所示.图1(a)为原始碳纳米管,可以看到其为直径在15-40 nm左右的多壁碳纳米管,且不含催化剂等杂质.图1(b-d)为聚苯胺/碳纳米管复合物PANI/CNT-1到PANI/CNT-3的形貌,可看出,碳管表面形成了一定厚度的包覆层,依据苯胺用量不同,包覆层厚度也有差异,随着苯胺用量的增加,层厚度增大.苯胺用量在0.15,0.30和0.60 g的聚苯胺/碳纳米管复合物样品的层厚度分别约为12、17和25 nm.为了获得氮掺杂碳纳米管,将聚苯胺/碳纳米管复合物在700°C下进行炭化处理,得到的样品NCNT-1、NCNT-2和NCNT-3的形貌见图1(e-f),可看出,聚苯胺/碳纳米管复合物经炭化处理后,仍保持纤维状形貌,且纤维状产物仍相互交织呈网络形态.炭化后,包覆层的厚度仍随苯胺用量的增大而增大,但对比聚苯胺/碳纳米管复合物,炭化后的碳管表面包覆层厚度减小,聚苯胺/碳纳米管复合物样品炭化后的膜厚度分别从12 nm降低到10 nm,17 nm减少至14 nm,25 nm减至20 nm,这可能是由于聚苯胺炭化后体积收缩的缘故.
3.2 氮掺杂处理前后碳纳米管的拉曼分析
图2为聚苯胺/碳纳米管复合物炭化处理前后(PANI/CNT-2和NCNT-2样品)及纯碳纳米管和聚苯胺的激光拉曼谱.对于碳纳米管,在波数为1330 cm-1的峰对应碳纳米管的D模,在1575和1605 cm-1的振动峰对应纳米碳管的G模.聚苯胺的拉曼峰位分别在1167 cm-1处对应琨基/苯基的C-H振动峰,1338、1359和1390 cm-1的肩峰对应C-N+·的拉伸振动,在1595 cm-1处是C-C振动峰,1479和1512 cm-1处是聚苯胺结构中掺杂态的特征峰.聚苯胺/碳纳米管复合物的拉曼谱同时显现出碳纳米管和聚苯胺的拉曼特征峰,因此,结合聚苯胺/碳纳米管复合物的TEM分析,可以确认复合物中碳纳米管表面包覆层为聚苯胺层.聚苯胺/碳纳米管复合物经炭化处理后,碳纳米管的特征峰与炭化前相比变得更明显,说明炭化处理没有破坏碳管的本征结构;进一步,炭化处理后,聚苯胺特征峰消失,表明碳管表面包覆的聚苯胺层完全转化为炭化层.

图1 原始CNTs(a),聚苯胺/碳纳米管复合物PANI/CNT-1(b)、PANI/CNT-2(c)、PANI/CNT-3(d)和氮掺杂处理后的样品NCNT-1(e)、NCNT-2(f)、NCNT-3(g)的透射电镜照片Fig.1 TEM images of pristine CNTs(a),composites of PANI/CNT-1(b),PANI/CNT-2(c),PANI/CNT-3(d)and N doping samples of NCNT-1(e),NCNT-2(f),NCNT-3(g)

图2 聚苯胺/碳纳米管复合物炭化前后及聚苯胺和原始碳纳米管的拉曼光谱Fig.2 Raman spectra of PANI/CNT composite before and after carbonization,PANI and the pristine CNT
3.3 氮掺杂前后碳纳米管的XPS分析
X射线光电子谱(XPS)被用于样品的表面化学组成分析,原始碳纳米管和经炭化处理的聚苯胺包覆碳纳米管(NCNT-2)的XPS见图3.从XPS全扫描图(图3(a))可看出,原始碳纳米管除含氧元素外,不含氮元素,而聚苯胺包覆碳纳米管经炭化处理后样品的XPS谱线中出现明显的N 1s峰,因此,结合经炭化处理的样品的透射电镜观察和拉曼分析结果,可以确认炭化处理使聚苯胺包覆碳纳米管形成了外部为氮掺杂碳层、内部为碳纳米管结构的氮掺杂碳纳米管.对不同苯胺用量制备的氮掺杂碳纳米管的XPS氮含量分析结果列于表1,可看出随着苯胺用量增加,氮掺杂碳纳米管不仅氮掺杂层厚度增大,且氮含量也略有增大.

表1 不同氮掺杂层厚度的碳纳米管的氮含量分析结果Table 1 Nitrogen content of NCNTs with the different thicknesses of N-doped layer
为了进一步分析氮掺杂碳层氮原子官能团信息,对NCNT-2样品的XPS N 1s谱进行了高斯拟合分峰处理,结果如图3(b)所示.可以看出,N 1s谱线通过分峰处理可得到4个峰位,分别位于397.9、399.8、400.6及402.0 eV,它们分别对应吡啶型氮(=N-)、吡咯型氮(-NH-)、取代型氮(N+)和氧化吡啶型氮(=N+-).12,23,27
3.4 氮掺杂碳纳米管的超级电容器特性
原始碳纳米管和氮掺杂碳纳米管电极在6 mol·L-1氢氧化钾电解液中的循环伏安(CV)曲线见图4.原始碳纳米管的CV曲线形近似为矩形,但在0.1 V处出现氧化还原峰,由于原始碳纳米管中不含氮元素,其可能是含氧官能团引起的准法拉第反应的结果.而氮掺杂碳纳米管的CV曲线呈面积更大的准矩形,暗示了氮掺杂处理使碳纳米管的电容明显增大.根据CV曲线计算得到的CNTs和NCNT-1、NCNT-2和NCNT-3的比容量分别为10、205、145和107 F·g-1,聚苯胺改性氮掺杂处理使碳纳米管电极的比电容量显著增强,增强的电容应来源于氮杂原子引入的法拉第电容和使碳纳米管亲水性改善而增加的双电层电容.进一步可看出,随着氮掺杂层厚度的增大,NCNTs的比电容明显降低,尽管更厚的氮掺杂层具有略高的氮掺杂量,其原因可能是:一方面,只有氮掺杂碳层外层的表面氮杂原子能够有效贡献法拉第电容,NCNT-1、NCNT-2和NCNT-3的比表面积分别为80.2、72.6和62.5 m2·g-1,相比之下氮掺杂层薄的样品(NCNT-1)具有最高的比表面积,从而能够提供更多的表面氮杂原子,即增强碳管的法拉第电容;另一方面,由于氮掺杂原子能够改善碳管亲水性从而增强双层电容,因此掺杂层薄的样品不仅具有更多的表面氮杂原子从而有效改善碳管亲水性,而且其碳纳米管相对含量更高,这些亲水性得到改善的碳纳米管也贡献了更多的双层电容,因此,氮掺杂层较薄的碳管具有更高的比电容.然而由于氮掺杂层和碳纳米管贡献电容的机理不同,目前还很难定量区分它们各自对电容量的贡献.
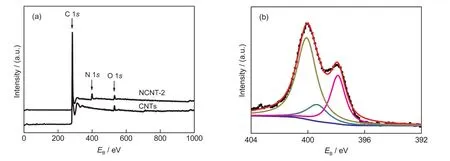
图3 氮掺杂处理前后碳纳米管的XPS扫描图谱(a)和氮掺杂碳纳米管的N 1s谱图(b)Fig.3 XPS survey scans of CNTs before and after N-doping(a)and N 1s core-level spectra of NCNTs(b)
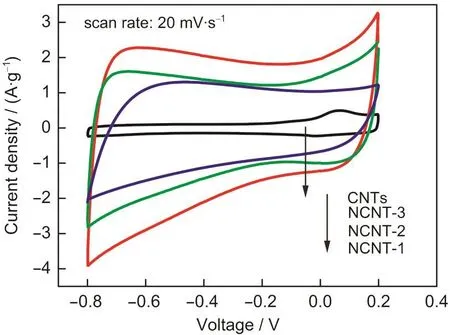
图4 原始碳纳米管和氮掺杂碳纳米管的循环伏安曲线Fig.4 Cyclic voltammograms of the pristine CNTs and NCNTs
基于苯胺改性制备的氮掺杂碳纳米管不仅具有高的比容量,而且也表现了较好的充放电循环性能,图5显示了氮掺杂纳米管比容量随充放电循环次数的变化.可看出,所有样品都具有较好的循环稳定性,经 1000次循环后,NCNT-1、NCNT-2和NCNT-3分别保持了其初始容量的92.8%、93.6%和97.1%,具有较厚氮掺杂层的碳纳米管的充放电循环稳定性更优,其可能是由于厚的氮掺杂层与基体碳纳米管结合更加稳定的缘故.氮掺杂碳纳米管作为超级电容器电极良好的循环性能够归因于其组织和结构特征,TEM观察和XPS分析表明,苯胺改性处理制备的氮掺杂碳纳米管具有以外氮掺杂碳层为壳、内碳纳米管为核的核-壳结构,氮掺杂碳层具有均一厚度,而且氮掺杂层源于聚苯胺包覆层的炭化,因此氮杂原子均匀分布于外部碳层;碳纳米管核作为良好的电子导体有利于法拉第过程的电荷传输;另一方面,其作为氮掺杂碳层支持体有利于缓解法拉第反应导致体积变化,因此,与氧化剂(如强酸或氨气)处理使含氮官能团主要形成于碳纳米管缺陷位,并导致碳纳米管本征结构特性遭受破坏而制备的氮掺杂碳纳米管相比,苯胺改性处理制备的具有核-壳结构特征的氮掺杂碳纳米作为超电极电容器电极表现出更好的循环稳定性.
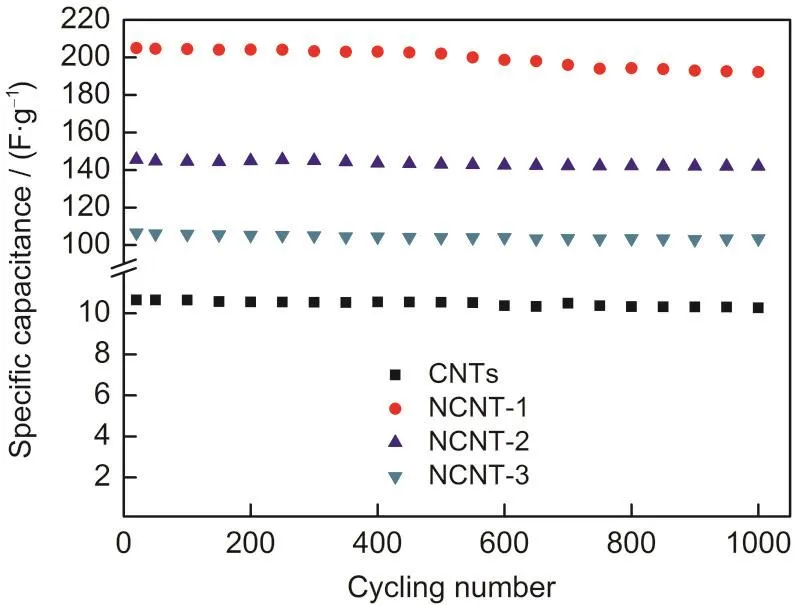
图5 原始碳纳米管和氮掺杂碳纳米管作为电容器电极材料的充放电循环寿命Fig.5 Charge-discharge cycling life of the pristine CNTs and NCNTs as electrode material of supercapacitor
4 结论
通过合成聚苯胺包覆碳纳米管,再对其进行炭化处理,使碳纳米管表面包覆的聚苯胺转化成氮掺杂碳层,从而成功制备出以碳纳米管为核,氮掺杂碳层为壳,具有核-壳结构特征的氮掺杂碳纳米管.并通过改变苯胺的用量,制备了具有不同氮掺杂层厚度的碳纳米管,其氮含量随氮掺杂层厚度增大而增大,从7.06%增加到8.64%.作为超级电容器电极,因表面氮杂原子引入的法拉第电容和使碳纳米管亲水性改善而提供的双电层电容,聚苯胺改性氮掺杂处理使碳纳米管比容量从10 F·g-1增大到107-205 F·g-1,且其电容量随氮掺杂层厚度降低而增大;尤其由于氮掺杂碳纳米管稳定的核-壳结构特征和碳纳米管表面均匀的氮掺杂层,其作为超级电容器电极也显示出了良好的循环性,经1000次充放电循环,聚苯胺改性处理制备的具有不同氮掺杂层厚度碳纳米管电极仍保持了初始容量的92.8%-97.1%.
(1) Dresselhaus,M.S.;Dresselhaus,G.;Eklund,P.C.Science of Fullerenes&Carbon Nanotubes;San Diego:Academic Press,March 1996;pp 20-35.
(2)Treacy,M.M.J.;Ebbesen,T.W.;Gibson,J.M.Nature 1996,381,678.doi:10.1038/381678a0
(3) Frackowiak,E.;Metenier,K.;Bertagna,V.;Beguin,F.Appl.Phys.Lett.2000,77,2421.doi:10.1063/1.1290146
(4)Li,C.S.;Wang,D.Z.;Wang,X.F.;Liang,J.Carbon 2005,43,249.
(5)Baughman,R.H.;Zakhidov,A.A.;de Heer,W.A.Science 2002,297,787.doi:10.1126/science.1060928
(6) Shiratori,Y.;Sugime,H.;Noda,S.J.Phys.Chem.C 2008,112,17974.
(7)Hou,P.X.;Orikasa,H.;Yamazaki,T.;Matsuoka,K.;Tomita,A.;Setoyama,N.;Fukushima,Y.;Kyotani,T.Chem.Mater.2005,1,5187.
(8) Eduardo,C.S.;Florentino,L.U.;Emilio,M.S.ACS Nano 2009,3,1913.doi:10.1021/nn900286h
(9)Yang,Y.;Li,X.;Jiang,J.;Du,H.;Zhao,L.;Zhao,Y.ACS.Nano 2010,4,5755.doi:10.1021/nn1014825
(10) Byrne,J.;Li,Z.;Jones,S.;Fleming,P.;Larsson,J.A.;Morris,M.A.;Holmes,J.D.ChemPhysChem.2011,12,2995.doi:10.1002/cphc.v12.16
(11) Liu,Y.;Jin,Z.;Wang,Y.;Cui,R.L.;Sun,H.;Peng,F.;Wei,L.;Wang,Z.X.;Liang,X.L.;Peng,L.M.;Li,Y.Adv.Funct.Mater.2011,21,986.doi:10.1002/adfm.201002086
(12) Hulicova-Jurcakova,D.;Kodama,M.;Shiraishi,S.;Hatori,H.;Zhu,Z.H.;Lu,G.Q.Adv.Funct.Mater.2009,19,1800.doi:10.1002/adfm.v19:11
(13) Inagakia,M.;Konno,H.;Tanaike,O.J.Power Sources 2010,195,7880.
(14)Lota,G.;Grzyb,B.;Machnikowsk,H.;Machnikowski,J.;Frackowiak,E.Chem Phys.Lett.2005,404,53.doi:10.1016/j.cplett.2005.01.074
(15) Zhai,Y.P.;Dou,Y.Q.;Zhao,D.Y.;Fulvio,P.F.;Mayes,R.T.;Dai,S.Adv.Mater.2011,23,4828.doi:10.1002/adma.v23.42
(16) Gong,K.;Du,F.;Xia,Z.;Durstock,M.;Dai,L.Science 2009,323,760.doi:10.1126/science.1168049
(17) Tang,Y.;Allen,B.L.;Kauffman,D.R.;Alexander,S.J.Am.Chem.Soc.2009,131,13200.doi:10.1021/ja904595t
(18)Yang,S.;Zhao,G.L.;Khosravi,E.J.Phys.Chem.C 2010,114,3371.
(19) Sen,R.;Satishkumar,B.C.;Govindaraj,A.;Harikumar,K.R.;Renganathan,M.K.;Rao,C.N.R.J.Mater.Chem.1997,7,2335.doi:10.1039/a705891h
(20)Shirazi,Y.;Tofighy,M.A.;Mohammadi,T.;Pak,A.Appl.Sur.Sci.2011,257,7359.doi:10.1016/j.apsusc.2011.03.146
(21) Liu,J.;Rinzler,A.G.;Dai,H.J.;Hafner,J.H.;Bradley,R.K.;Boul,P.J.;Lu,A.;Iverson,T.;Shelimov,K.;Huffman,C.B.;Rodriguez-Macias,F.;Shon,Y.S.;Lee,T.R.;Colbert,D.T.;Smalley,R.E.Science 1998,280,1253.doi:10.1126/science.280.5367.1253
(22)Hiura,H.;Ebbesen,T.W.;Tanigaki,K.Adv.Mater.1995,7,275.
(23)Arrigo,R.;Havecker,M.;Schlogl,R.;Su,D.S.Chem.Commun.2008,4891.
(24) Halder,A.;Sharma,S.;Hegde,M.S.;Ravishankar,N.J.Phys.Chem.C 2009,113,1466.doi:10.1021/jp8072574
(25) Tessonnier,J.P.;Rosenthal,D.;Girgsdies,F.;Amadou,J.;Begin,D.;Pham-Huu,C.;Su,D.S.;Schlogl,R.Chem.Commun.2009,7158.
(26) Li,L.X.;Liu,Y.C.;Geng,X.;An,B.G.Acta Phys.-Chim.Sin.2011,27,443.[李莉香,刘永长,耿 新,安百刚.物理化学学报,2011,27,443.]doi:10.3866/PKU.WHXB20110225
(27)Niwa,H.;Horiba,K.;Harada,Y.;Oshima,M.;Ikeda,T.;Terakura,K.;Ozaki,J.I.;Miyata,S.J.Power Sources 2009,187,93.doi:10.1016/j.jpowsour.2008.10.064
