Characterization of 12 polymorphic microsatellite markers in the Chinese tree shrew (Tupaia belangeri chinensis)
2013-09-20XiaoHongLIUYongGangYAO
Xiao-Hong LIU , Yong-Gang YAO
1. Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology,Kunming, Yunnan 650223, China;
2. Kunming Primate Research Center, Kunming Institute of Zoology, the Chinese Academy of Sciences, Kunming 650223, China;
3. Tree Shrew Inbreeding Center, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China;
4. University of the Chinese Academy of Sciences, Beijing 100049, China
The Chinese tree shrew (Tupaia belangeri chinensis)is a squirrel-like animal. It belongs to the Family Tupaiidae of Scandentia. Tree shrews have the highest brain-to-body mass ratio of known mammals (Peng et al,1991). Because tree shrews share some characteristics with primates and insectivores, the exact phylogenetic position of the tree shrew has been debated (Arnason et al, 2002; Janecka et al, 2007; Peng et al, 1991; Xu et al,2012; Xu et al, 2013a). Analyses of the mitochondrial DNA (mtDNA) genome have shown that the tree shrew has a closer affinity with Lagomorpha (Arnason et al,2002; Xu et al, 2012), while nuclear gene sequences provide evidence for a close affinity to primates (Janecka et al, 2007; Killian et al, 2001; Lindblad-Toh et al, 2011).
The tree shrew has long been proposed to be an alternative experimental animal to primates in biomedical research due to its characteristics such as small body size, short reproductive cycle and life span,and low-cost maintenance (Peng et al, 1991). This animal has been used to create animal models for infection with the hepatitis B and C viruses, the development of hepatocellular carcinoma, myopia, psychosocial stress(Cao et al, 2003; McBrien & Norton, 1992; van Kampen et al, 2002; Yan et al, 1996a; Yan et al, 1996b) and learning and memory (Wang et al, 2011), although there are many problems awaiting further studies (Xu et al,2013b) . Despite an increasing interest in using the tree shrew to establish animal models for medical and biological research, most of the animals used were captured from the wild and/or domesticated for a few months. There is no pure inbreeding strain with a clear genetic background similar to that of a mouse or rat at the present time, which would largely influence the stability and repeatability of experiments.1
Tupaiidae contains four genera: Tupaia, Anathana,Urogale, and Dendrogale (Helgen, 2005). Currently, a total of 15 species is recognized in Tupaia, which is broadly distributed across South and Southeast Asia,including southern China, India, Philippines, Java,Borneo, Sumatra and Bali (Olson et al, 2005; Peng et al,1991). Chinese tree shrews (Tupaia belangeri) are distributed across southwest China and are divided into six subspecies according to geographical distribution and morphological characteristics (Wang, 1987).Understanding the genetic diversity of the wild tree shrew population has critical importance for conservation and for a breeding program to establish inbred lines. Recently, we evaluated the mtDNA genetic diversity of the tree shrew inhabiting urban Kunming and observed a relatively high diversity (Chen et al, 2011).Genetic markers from the nuclear genome are essential for us to further validate this conclusion and to analyze other behaviors of this animal, e.g., female-biased dispersal and gene flow between different populations.
Microsatellites are short tandem repeats (STR) in the genome and have been broadly used for evaluating genetic diversity, population affinity, gene flow and population microdifferentiation (Goldstein et al, 1999;Schlöetterer & Pemberton, 1994; Waits et al, 2000), as well as individual identification and parentage tests (Behl et al, 2002; Butler, 2005). So far, there have been only sporadic reports for characterizing microstatellites in the tree shrew (Munshi-South, 2008; Munshi-South &Wilkinson, 2006; Srikwan et al, 2002). Amplification of the Chinese tree shrew genomic DNA using microsatellites developed from other species of Scandentia revealed some problems, such as lack of polymorphism (He et al, 2009) and had non-specific amplification in our samples (data not shown). Therefore,it is necessary to develop a set of species-specific microsatellite markers based on the available genome of the tree shrew. In this study, we successfully retrieved 12 polymorphic microsatellite markers from Chinese tree shrews and characterized them in a population of 117 individuals. Our results showed that this set of microsatellite loci had sufficient data to measure population heterozygosity and individual discrimination.
MATERIALS AND METHODS
Experimental animals
A total of 117 Chinese tree shrews inhabiting the suburb of Kunming, Yunnan Province, China, were collected and raised at the Experimental Animal Core Facility of the Kunming Institute of Zoology, Chinese Academy of Sciences. All sampling procedures were approved by the Institute’s Institutional Animal Care and Use Committee. Genomic DNA was extracted from the blood, ear and/or muscle tissues of each individual by using the genomic DNA extraction kit (Tiangen, Beijing).
Microsatellite identification and amplification
Based on the genomic DNA sequences of the northern tree shrew (Tupaia belangeri) provided by the Ensembl database (ftp://ftp.ensembl.org/), 20 loci were chosen with a core repeat unit of 4-6 nucleotides. Primer pairs were designed using primer premier 5. PCR amplification condition for each locus contained a predenaturation cycle at 94 ℃ for 5 min, followed by 30 cycles of denaturation at 94 ℃ for 30 s, annealing at 55 ℃ for 30 s, and extension at 72 ℃ for 30 s, followed by a final extension cycle at 72℃ for 5 min.Amplification was performed in a total volume of 20 μL,which contained 10×Taq polymerase reaction buffer(100 mmol/L Tris-HCl, pH 8.3; 500 mmol/L KCl; 15 mmol/L MgCl2), 0.2 mmol/L dNTPs, 10 μmol/L of each forward and reverse primer, 0.5 U Taq DNA polymerase(TaKaRa, Dalian, China), and 50 ng genomic DNA. PCR products were electrophoresed on 8% polyacrylamide gels to test successful and specific amplification. For those loci with no amplification or with non-specific bands, we optimized the reaction conditions by increasing or decreasing the annealing temperature, or adjusting the concentration of Mg2+to achieve the best efficiency and specificity. We validated the authentic sequencing of each amplified locus by TA-cloning sequencing. In brief, PCR product was ligated into PMD 19-T vector (TaKaRa, Dalian, China), followed by transforming DH5α competent cells (Tiangen, Beijing)according to the manufacturer’s instructions. We picked up three to five plasmids with the right inserts for sequencing.
After the pretest for primer pairs and the optimization for amplification, we labeled the forward primer of each locus by including 6-FAM (Carboxyfluorescein) at the 5' end of the primer. PCR amplification for each locus was performed using the above optimized conditions. PCR product was diluted with double-distilled water to achieve a concentration of 1 ng/μL. We mixed 1 μL diluted PCR product with 0.1 μL GenescanTM-500 LIZ®(Applied Biosystems) and Hi-DiTMFormamide (Applied Biosystems), and added ddH2O to make a 10 μL volume of cocktail. The mixture was denatured at 95 ℃ for 5 min, then immediately chilled on ice. We loaded 1 μL of cocktail on an automated sequencer (ABI PRISM 3100, Applied Biosystems). Alleles were scored using program Genemarker V1.5 (SoftGenetics LLC, State College, PA).
Statistical analysis
Allele frequencies, observed heterozygosity (Ho),expected heterozygosity (He) and deviation from the Hardy-Weinberg equilibrium (HWE) based on the likelihood ratio test were calculated by POPgene 1.32(Yeh & Boyle, 1997). Polymorphism information content(PIC), power of discrimination (PD), power of exclusion(probability of excluding relatives of the true father from paternity, PE) and paternity index (PI, which is the ratio of the probability that a genetic marker/allele the alleged father passed to the child to the probability that a randomly selected unrelated man of similar ethnic background could pass the allele to the child) were calculated by PowerStats V1.2 (Promega, Madison, WI).Non-exclusion probabilities (NEP), probability of identity (Pid), probability of identity of siblings (Pid-sib)and null allele frequency (F (null)) were calculated using Cervus 3.0 (Kalinowski et al, 2007).
RESULTS
Characterization of the microsatellites
Among the 20 selected loci, only 14 could be successfully amplified in the Chinese tree shrew using the primer pairs designed in this study (Table 1). The sequence authenticity of each locus was verified by sequencing and the microsatellite sequences were deposited in GenBank under accession numbers JQ173849-JQ173882 (Figure 1). There are some mutations in the repeat motif (e.g., from AAG to GAG in loci TB9 and TB14), besides the change of the number of repeats. The remaining 6 loci either failed to be amplified or with apparent non-specific amplification (Table S1).
To test whether the 14 loci were polymorphic in the Chinese tree shrew population, we screened 117 individuals by using 5'-fluorescently labeled primers.Two loci, TB2 and TB10, were non-polymorphic in the tested samples. However, a comparison of the sequencedfragments of these two loci with the reference sequence from the Ensembl database showed length polymorphisms (Figure S1), suggesting that these two loci might present polymorphisms among tree shrews from different regions, but not in tree shrews collected from Kunming. This result was in accordance with our previous studies of mtDNA sequence variation in the Chinese tree shrew (Chen et al, 2011; Xu et al, 2012), in which we observed some nucleotide differences between the Chinese tree shrew from Kunming and the reported northern tree shrew outside China, though both were grouped as Tupaia belangeri.

Table 1 Information of 14 primer pairs for amplifying microsatellite loci in Chinese tree shrew(Tupaia belangeri chinensis) and PCR condition
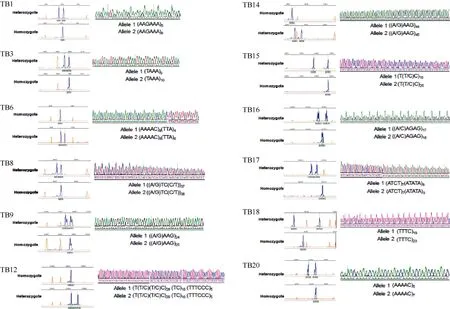
Figure 1 GeneScan maps and sequences of 12 polymorphic microsatellites in Chinese tree shrew population
Chinese tree shrew population has a high microsatellite polymorphism
Tables 2 and 3 list the estimated parameters for each of the 12 polymorphic microsatellites in Chinese tree shrews. The number of alleles per locus ranged from 4 to 20, with an average of 8.75 alleles. The effective alleles ranged from 1.19 (TB1 locus) to 10.47 (TB18 locus), and had an average of 4.05. The mean values of observed heterozygosity and expected heterozygosity were 0.566 and 0.616, respectively. Loci TB1 and TB15 had a relatively low level of polymorphism (PIC value<0.25),TB3 and TB6 were moderately polymorphic (0.25<PIC value<0.5), and the other loci were highly polymorphic(PIC value>0.5) (Table 2). The high PIC value and observed heterozygosity indicated a high genetic diversity of the analyzed tree shrew population, whichwas consistent with the previous mtDNA analysis (Chen et al, 2011). A likelihood ratio test for concordance with HWE revealed that only loci TB12, TB17 and TB20 deviated from HWE (P<0.05), but none was significant after applying a Bonferroni correction.

Table 2 Genetic diversity measures for 12 polymorphic microsatellite loci tested in 117 Chinese tree shrews
To further evaluate the potential use of the 12 microsatellite markers, we calculated some forensic efficiency parameters based on the tested samples. The linkage disequilibrium was tested by the online SHEsis program (Shi & He, 2005), but no significant linkage between loci was detected (Table S2). The power of discrimination (PD) was from 0.218 (TB1) to 0.971(TB18), and the cumulative PD in this study was as high as 99.99999998%. Waits et al (2001) recommend a reasonable probability of identifying individuals in a given region using dominant and codominant markers as 0.01-0.0001. In this study, the values of combined Pid and combined Pid-sib were 4.89×10-11and 0.00001,respectively. These values are far smaller than 0.01,which suggests that these 12 loci had sufficient data for individual identification.
As testing for more markers would increase the cost,time and the risk of analytical errors (Waits & Paetkau,2005), it was necessary to choose the minimum number of markers that fulfill the aim of individual identification.We eliminated the four less polymorphic loci (TB6, TB3,TB15 and TB1) but still obtained considerably high values for combined PD (CPD=99.99999955%),combined Pid (CPid=8.61×10-10), and combined Pid-sib(CPid-sib=5.28×10-5). It is thus evident that the remaining 8 loci are sufficient for individual discrimination.
For parentage testing, parameters such as average non-exclusion probability (NE-1P), average nonexclusion probability for one candidate parent given the genotype of a known parent of the opposite sex (NE-2P),average non-exclusion probability for a candidate parent pair (NE-PP), power of exclusion (PE) and paternity index (PI) were widely used in the field. The Chinese tree shrew population had a NE-1P value of 0.322-0.994,a NE-2P value of 0.192-0.945, a NE-PP value of 0.059-0.897, a PE value of 0.668-0.012, and a PI value of 3.05-0.57, respectively (Table 3). Excluding the four loci (TB3, TB15, TB1, and TB6) with few polymorphisms, we still obtained sufficient data for parentage testing.
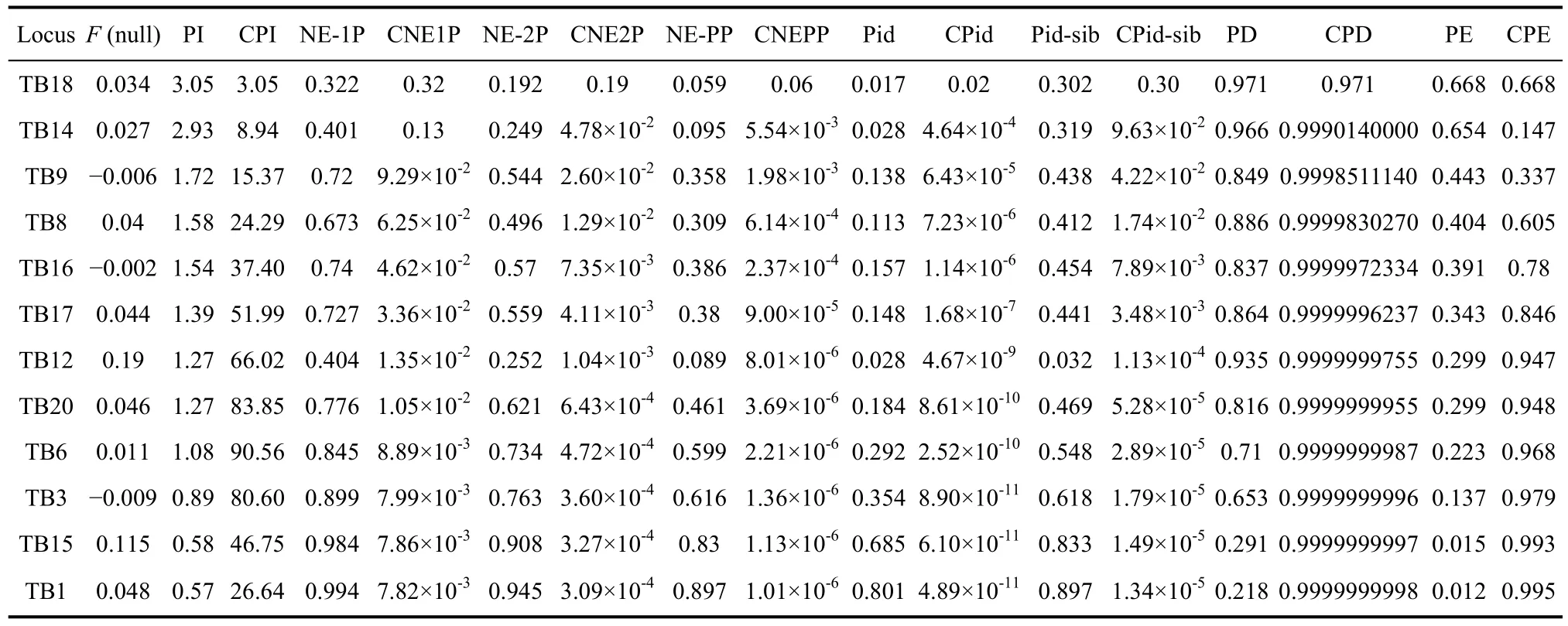
Table 3 Individual identification and parentage testing parameters for 12 polymorphic microsatellite markers in a population of Chinese tree shrew (n=117)
The null allele frequency was also calculated to evaluate the efficiency of this set of microsatellite markers because of its influence on population analysis (Chapuis& Estoup, 2007). Null allele is any microsatellite allele at a microsatellite locus that consistently fails to amplify to detected levels in the PCR assays, and it has no significant effect on parentage analysis when the frequency is less than 0.2 (Dakin & Avise, 2004). In our study, all the null allele frequencies were less than 0.2 (Table 3). Therefore,these markers can be used for population analysis of tree shrews, at least in parentage analysis with little influence on the average exclusion probability.
DISCCUSSION
Tree shrews have received wide attention in recent years for their potential use in biomedical research and drug discovery (Cao et al, 2003; Fuchs & Corbach-Söhle,2010; Peng et al, 1991). Despite decades of cultivation efforts, no inbred strain has been established for the tree shrew, which hindered a wide use of this animal in biomedical research; this might account for the inconsistency of previous results concerning animal models. Thus establishing some inbred lines of tree shrews is crucial, and we have launched an ambitious project to fulfill this goal.
To facilitate our current inbreeding program for the Chinese tree shrew and to evaluate the genetic diversity of wild tree shrews, we needed a set of microsatellite markers that had sufficient data for individual discrimination and parentage testing, as well as for the quantification of population heterogeneity. In this study,by referring to the available tree shrew genome sequence(albeit the coverage was only 2×), we were able to obtain 12 polymorphic loci in the Chinese tree shrew.Evaluation of different parameters related to genetic diversity, individual identification and parentage testing showed that eight of these loci had sufficient data for lineage tracing and measurement of diversity.
Compared with these reported microsatellite markers, which were developed for T. glias and T. minor(Munshi-South & Wilkinson, 2006; Srikwan et al, 2002),our newly designed set based on the genome sequence of T. belangeri was specifically optimized for Chinese tree shrews. As we had no samples of other species in Tupaia,we did not know whether our primer pairs would work for these species. As Chinese tree shrews were proposed to contain six subspecies (Wang, 1987), it would be rewarding to screen the genetic diversity of these different subspecies, both for conservation of genetic resources and for selecting a proper founder to establish the inbred lines.
Microsatellite markers have long been used in genetic analyses for both wild populations and/or domestic animals. In general, wild populations have a higher genetic diversity compared to the inbred strain or domestic breeds. Analysis of the inbred C57BL/6J mouse strain using microsatellite markers revealed a lower expected heterozygosity (<0.5) (Niu & Liang,2009),but the expected heterozygosity of the wild house mouse(Mus musculus) reached 0.62 in population from Cameroon and 0.82 in population from Germany(Thomas et al, 2005). Based on the analyses of 19 microsatellite loci, three pure breeds of dog (Greyhounds,Labradors and German Shepherds) showed expected low heterozygosities of 0.357, 0.481 and 0.431, respectively(Zajc et al, 1997). In this study, the expected heterozygosity of the Chinese tree shrew was 0.616,lower than that of the plain treeshrew (T. longipes) (0.74)but higher than that of the large treeshrew (T. tana) (0.58)distributed in Sabah, Malaysia (Munshi-South &Wilkinson, 2006). As our population size was considerably large (n=117), we speculated that the observed heterozygosity might reflect the true situation of wild tree shrews distributed in Kunming, Yunnan Province.
In short, we characterized a set of 12 polymorphic microsatellite markers for the Chinese tree shrew, which were confirmed to be sufficient for assessing population genetic structure and individual discrimination of this animal. An analysis of 117 wild individuals showed that the Chinese tree shrew had a considerably high heterozygosity. We hope that these markers will provide essential help for us to advance the inbreeding project of the Chinese tree shrew.
Acknowledgements: We thank Mr. Long FAN in YAO’s lab for assistance with data analysis.
Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A,Nilsson M, Short RV, Xu X, Janke A. 2002. Mammalian mitogenomic relationships and the root of the eutherian tree. Proc Natl Acad Sci USA,99(12): 8151-8156.
Behl R, Sheoran N, Behl J, Tantia MS, Vijh RK. 2002. Microsatellite sequences of mammals and their applications in genome analysis in pigs: A review. Asian Austral J Anim, 15(12): 1822-1830.
Butler JM. 2005. Forensic DNA Typing: Biology, Technology and Genetics of STR Markers. New York: Elsevier Academic Press.
Cao J, Yang EB, Su JJ, Li Y, Chow P. 2003. The tree shrews: adjuncts and alternatives to primates as models for biomedical research. J Med Primatol, 32(3): 123-130.
Chapuis MP, Estoup A. 2007. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol, 24(3): 621-631.
Chen SY, Xu L, Lu LB, Yao YG. 2011. Genetic diversity and matrilineal structure in Chinese tree shrews inhabiting Kunming, China.Zool Res, 32(1): 17-23.
Dakin EE, Avise JC. 2004. Microsatellite null alleles in parentage analysis. Heredity, 93(5): 504-509.
Fuchs E, Corbach-Söhle S. 2010. Tree Shrews // The UFAW Handbook on the Care and Management of Laboratory and Other Research Animals. Oxford: Wiley-Blackwell, 262-275.
Goldstein DB, Roemer GW, Smith DA, Reich DE, Bergman A, Wayne RK. 1999. The use of microsatellite variation to infer population structure and demographic history in a natural model system. Genetics,151(2): 797-801.
He BL, Shen PQ, Chen LL, Jiao JL, Liu RW, Li B, Zheng H, Li ML.2009. Polymorphism microsatellites in tree shrews (Tupaia belangeri chinensis). Acta Lab Anim Sci Sin, 17(2): 143-145 (in Chinese).
Helgen KM. 2005. Order Scandentia // Wilson DE, Reeder DM.
Mammal Species of the World: A Taxonomic and Geographic Reference. Maryland: Johns Hopkins University Press, 104-109.
Janecka JE, Miller W, Pringle TH, Wiens F, Zitzmann A, Helgen KM,Springer MS, Murphy WJ. 2007. Molecular and genomic data identify the closest living relative of primates. Science, 318(5851): 792-794.
Kalinowski ST, Taper ML, Marshall TC. 2007. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol, 16(5): 1099-1106.
Killian JK, Buckley TR, Stewart N, Munday BL, Jirtle RL. 2001.Marsupials and Eutherians reunited: genetic evidence for the Theria hypothesis of mammalian evolution. Mamm Genome, 12(7): 513-517.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S,Kheradpour P, Ernst J, Jordan G, Mauceli E, Ward LD, Lowe CB,Holloway AK, Clamp M, Gnerre S, Alfoldi J, Beal K, Chang J,Clawson H, Cuff J, Di Palma F, Fitzgerald S, Flicek P, Guttman M,Hubisz MJ, Jaffe DB, Jungreis I, Kent WJ, Kostka D, Lara M, Martins AL, Massingham T, Moltke I, Raney BJ, Rasmussen MD, Robinson J,Stark A, Vilella AJ, Wen J, Xie X, Zody MC, Baldwin J, Bloom T,Chin CW, Heiman D, Nicol R, Nusbaum C, Young S, Wilkinson J,Worley KC, Kovar CL, Muzny DM, Gibbs RA, Cree A, Dihn HH,Fowler G, Jhangiani S, Joshi V, Lee S, Lewis LR, Nazareth LV,Okwuonu G, Santibanez J, Warren WC, Mardis ER, Weinstock GM,Wilson RK, Delehaunty K, Dooling D, Fronik C, Fulton L, Fulton B,Graves T, Minx P, Sodergren E, Birney E, Margulies EH, Herrero J,Green ED, Haussler D, Siepel A, Goldman N, Pollard KS, Pedersen JS,Lander ES, Kellis M. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature, 478(7370): 476-482.
McBrien NA, Norton TT. 1992. The development of experimental myopia and ocular component dimensions in monocularly lid-sutured tree shrews (Tupaia belangeri). Vision Res, 32(5): 843-852.
Munshi-South J, Harpending H. 2008. Female-biased dispersal and gene flow in a behaviorally monogamous mammal, the large treeshrew(Tupaia tana). PLoS One, 3(9): e3228.
Munshi-South J, Wilkinson GS. 2006. Isolation and characterization of polymorphic microsatellite loci in Bornean treeshrews (Tupaia spp.).Mol Ecol Notes, 6(3): 698-699.
Niu Y, Liang S. 2009. Genetic differentiation within the inbred C57BL/6J mouse strain. J Zool, 278(1): 42-47.
Olson LE, Sargis EJ, Martin RD. 2005. Intraordinal phylogenetics of treeshrews (Mammalia: Scandentia) based on evidence from the mitochondrial 12S rRNA gene. Mol Phylogenet Evol, 35(3): 656-673.Peng YZ, Ye ZZ, Zou RJ, Wang YX, Tian BP, Ma YY, Shi LM. 1991.Biology of Chinese Tree Shrews (Tupaia belangeri chinensis).Kunming, China: Yunnan Science and Technology Press.
Schlöetterer C, Pemberton J. 1994. The use of microsatellites for genetic analysis of natural populations. – In: Schierwater B, Streit B,Wagner GP & DeSalle R (Eds). Molecular ecology and evolution:approaches and applications. Experientia Supplementum 69.Birkhäuser Verlag Basel, Switzerland, pp. 203-214
Shi YY, He L. 2005. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res, 15(2): 97-98.
Srikwan S, Hufford K, Eggerf L, Woodruff DS. 2002. Variable microsatellite markers for genotyping tree shrew, tupaia, and their potential use in genetic studies of fragmented populations. Science Asia,28(2): 93-97.
Thomas M, Ihle S, Ravaoarimanana I, Kraechter S, Wiehe T, Tautz D.2005. Microsatellite variability in wild populations of the house mouse is not influenced by differences in chromosomal recombination rates.Biol J Linn Soc, 84(3): 629-635.
van Kampen M, Kramer M, Hiemke C, Flugge G, Fuchs E. 2002. The chronic psychosocial stress paradigm in male tree shrews: evaluation of a novel animal model for depressive disorders. Stress, 5(1): 37-46.
Waits L, Taberlet P, Swenson JE, Sandegren F, Franzen R. 2000.Nuclear DNA microsatellite analysis of genetic diversity and gene flow in the Scandinavian brown bear (Ursus arctos). Mol Ecol, 9(4): 421-431.
Waits LP, Paetkau D. 2005. Noninvasive genetic sampling tools for wildlife biologists: A review of applications and recommendations for accurate data collection. J Wildlife Manage, 69(4): 1419-1433.
Waits LP, Luikart G, Taberlet P. 2001. Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol, 10(1): 249-256.
Wang J, Zhou QX, Tian M, Yang YX, Xu L. 2011. Tree shrew models:a chronic social defeat model of depression and a one-trial captive conditioning model of learning and memory. Zool Res, 32(1): 24-30.
Wang YX. 1987. Taxonomic research on Burma-Chinese tree shrew,Tupaia belangeri (Wagner), from Southern China. Zool Res, 8(3): 213-230 (in Chinese).
Xu L, Chen SY, Nie WH, Jiang XL, Yao YG. 2012. Evaluating the phylogenetic position of Chinese tree shrew (Tupaia belangeri chinensis) based on complete mitochondrial genome: implication for using tree shrew as an alternative experimental animal to primates in biomedical research. J Genet Genomics, 39(3): 131-137.
Xu L, Fan Y, Jiang XL, Yao YG. 2013a. Molecular evidence on the phylogenetic position of tree shrew (Tupia belangeri). Zool Res, 34(2):70-76.
Xu L, Zhang Y, Liang B, Lv LB, Chen CS, Chen Y-B, Zhou JM, Yao YG. 2013b. Tree shrew under the spot light: emerging model of human diseases. Zool Res, 34 (2):59-69.
Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. 1996a.Human hepatitis B virus and hepatocellular carcinoma. I. Experimental infection of tree shrews with hepatitis B virus. J Cancer Res Clin Oncol,122(5): 283-288.
Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. 1996b.Human hepatitis B virus and hepatocellular carcinoma. II.Experimental induction of hepatocellular carcinoma in tree shrews exposed to hepatitis B virus and aflatoxin B1. J Cancer Res Clin Oncol,122(5): 289-295.
Yeh FC, Boyle TJB. 1997. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belg J Bot, 129(2): 157.
Zajc I, Mellersh CS, Sampson J. 1997. Variability of canine
microsatellites within and between different dog breeds. Mamm
Genome, 8(3): 182-185.
