Comparative systems biology between human and animal models based on next-generation sequencing methods
2013-09-20YuQiZHAOGongHuaLIJingFeiHUANG
Yu-Qi ZHAO, Gong-Hua LI, Jing-Fei HUANG,2,*
1. State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, the Chinese Academy of Sciences, Kunming Yunnan 650223,China;
2. Kunming Institute of Zoology, Chinese University of Hong Kong Joint Research Center for Bio-resources and Human Disease Mechanisms, Kunming Yunnan 650223, China
Accurately modeling the physiology and pathology of human systems research requires the establishment of a quality animal model (Alvarado & Tsonis 2006;Francia et al, 2011, 2006; Götz & Lttner 2008; Hasenfuss 1998; Lieschke & Currie, 2007). To this end, generally,how closely the model should mimic the human disease depends on the scientific question under investigation.Only in cases when the causal connections—structure function relationship or regulation of gene expression—are definitive, can the differences between human and animal models have minor effect on the analysis results(Hasenfuss, 1998). For example, although the zebrafish(Danio rerio) is phylogenetically distant from humans,its use as a complete animal model for in vivo drug discovery and development is growing rapidly(Chakraborty et al, 2009). However, if the pathophysiological processes are studied, especially for the complex diseases, then models should mimic clinical settings as closely as possible, otherwise the expected results may not be achieved or the findings of such studies will be of limited value.
Accordingly, comparisons between human and animal models are becoming increasingly important for both clinical and fundamental applications (Alini et al,2008; Cox et al, 2009; Fuentes et al, 2009; Huh et al,2010; Merchenthaler & Shughrue, 2005; Nestler &Hyman, 2010; Northoff, 2009). Among the available strategies to assess this connection, comparative systems biology has begun attracting special attention (Cox et al,2009).1
In this review, we introduce the concept of comparative systems biology. Next, we focus on the applications of next-generation sequencing methods,including RNA-seq and ChIP-seq, to comparative systems biology between human and animal models,before outlining some general directions of future developments and impacts of these types of studies.
The rise of comparative systems biology
One of the greatest twentieth century achievements in biological research is undoubtedly the sequencing of different genomes. There are now complete genome sequences for more than 1,000 organisms (excluding bacteria and archaea), with more sequences being completed (Henkelman, 2010). Once the genome of a species is available, researchers are able to begin mapping sequences against humans and find candidate disease genes and build a proper disease model. However,the ability to fundamentally understand the genotype–phenotype relationship in a distinct species is often hindered by the inherent complexity of biological systems. The difference in genotype–phenotype relationships between human and animal models may originate from three sources (Figure 1): (1) functional divergence of genes or proteins; (2) gene deletions or duplications; and (3) divergent up- or down-stream components, out of which gene deletions or duplications may play the leading role (Jaillon et al, 2004).
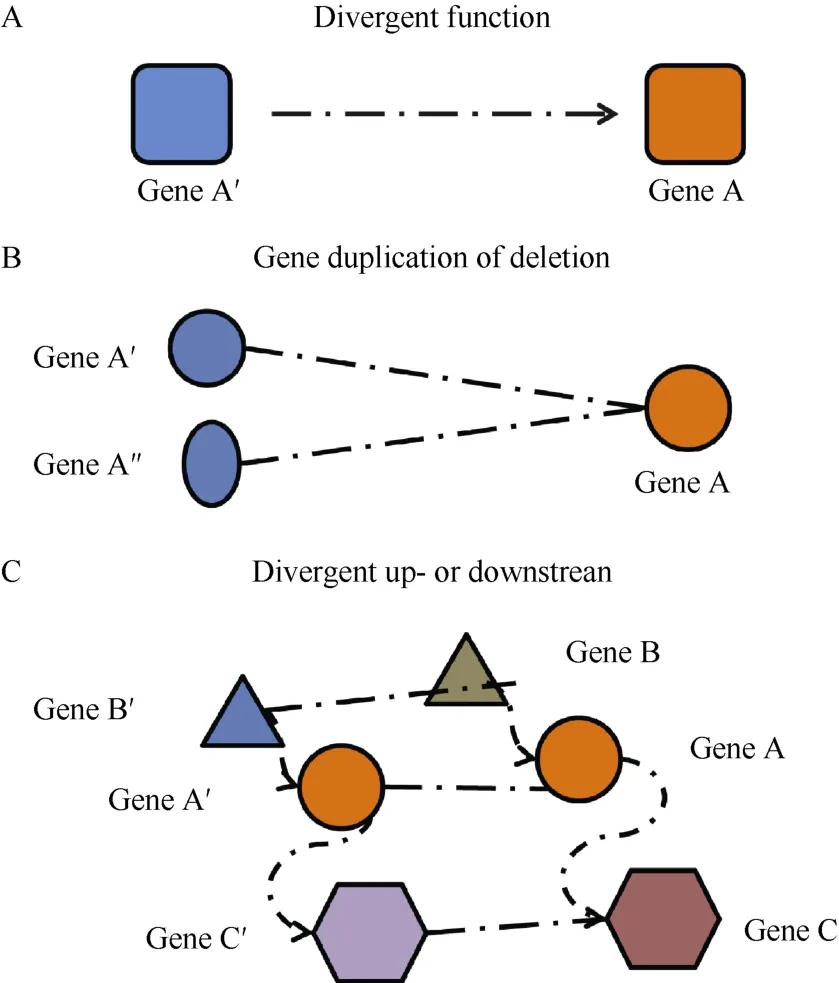
Figure 1 Mechanisms of different genotype-phenotype relationships between human and animal models
Over the last decade, this third mechanism has received more attention in systems biology. The Rb(Retinoblastoma) gene family is a good case, because the members in this family are functionally conserved while the involved pathways are divergent between C. elegans and humans (van den Heuvel & Dyson, 2008). Likewise,a previous study reported that over 20% of the essential genes for humans are non-essential for mice (Liao &Zhang, 2008). Consequently, traditional molecular biology techniques, while providing valuable insights into individual and/or simple genotype–phenotype relationship, are insufficient in deducing the complex phenotype-genotype relationships. Therefore, the more systematic methods at the systems biology level are necessary.
The ultimate goal of systems biology is generating successful models to comprehensively describe living organisms. Comparative systems biology, an important subfield of systems biology, has no straightforward definition. In animal model research, the term first appeared in Ogawa et al’s (2008) work, reporting a comparative study of circadian oscillatory network models of Drosophila. Here, we define comparative systems biology as “comparisons of biological systems in different states or species to achieve an integrated understanding of life forms with all their characteristic complexity of interactions at multiple levels.” The comparison can be performed either horizontally (e.g.,between individuals or states) or longitudinally (between species). The latter, which is mainly focused on human and animal models, is reviewed in detail here.
Over the past decade, comparative systems biology has attracted widespread interest, especially for its utility in comparisons between human and animal models of complex diseases. Miller et al (2010) used a systems biology approach to find a number of divergent network modules relevant to Alzheimer disease between humans and mice. In a previous work, we compared humans and four common animal models of cardiovascular disease through comparative transcriptome and pathway analysis,revealing that a few pathways have functionally diverged(Zhao et al, 2012). A recent review highlighted that the emerging technologies in comparative systems biology between human and animal models offers a platform to systematically explore not only the molecular mechanism of a particular disease, thus leading to the identification of disease modules and pathways, but also the molecular relationships among distinct(patho)phenotypes (Barabasi et al, 2011).
The majority of recent comparative systems biology studies on obtain their data through traditional high throughput technologies, such as microarray and ChIP-chip. Despite the experimental and statistical rigor as well as substantial insights gained through these methods,there has been a fundamental shift from these first-generation technologies (microarray and ChIP-chip) to next-generation sequencing (RNA-seq and ChIP-seq)over the last five years. We surmise that the applications of next-generation sequencing methods will serve a crucial function in the field of comparative systems biology between human and animal models, offering a number of potential advantages.
RNA-seq in transcriptome studies
Previous studies demonstrated that changes in gene expression underlie many or even most of phenotypic differences between species (Marques et al, 2008; Yanai et al, 2004). As a result, comparative transcriptome analysis potentially provides information on functional conservation for candidate human disease genes within animal models.
Initial trancriptomics studies largely relied on hybridization-based microarray technologies and have yielded valuable insights into the functional divergence between human and model animals (Enard et al, 2002;Liao & Zhang 2006). However, microarray technology has several limitations: over reliance upon existing knowledge about genome sequences; high background levels owing to cross-hybridization; and a limited dynamic range of detection owing to both background and saturation of signals (Wang et al, 2009). Recent advances in the DNA sequencing technology have enabled sequencing of cDNA derived from cellular RNA by massively parallel sequencing strategies, a process termed RNA-seq (Garber et al, 2011; Mortazavi et al,2008). Compared with the microarray, RNA-seq has the advantage of allowing high-resolution characterization and quantification of transcriptomes with low background noise and the ability to distinguish different isoforms.
Figure 2 shows the key procedures performed during RNA-seq analysis of comparative transcriptomes between human and animal models. The computational challenges in this process have been reviewed in detail by (Garber et al, 2011), therefore, we mainly illustrated the potential advantages of RNA-seq in comparative systems biology, including (a) comparisons between human and non-model animals, and (b) actual biological systems induced by the states of gene expression.
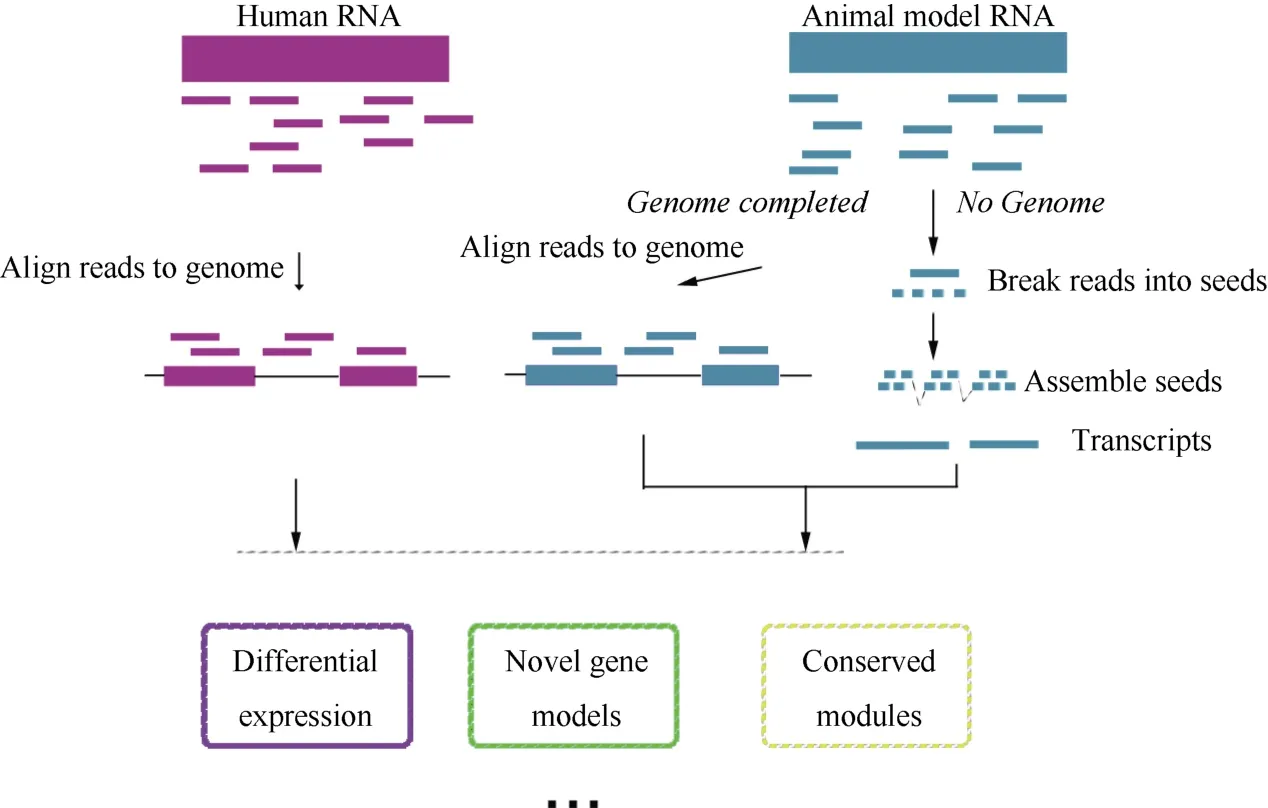
Figure 2 RNA-seq methods in comparative transcriptome analysisThere are two strategies for sequencing animal models. If the genome was not complete or was badly annotated, the genome-independent approach should be used (right part). The genome-guided approach is more typical (left part).
Though a variety of organisms have been genomically sequenced, the majority of these are used as model organisms. Since microarray relies on the genome information, this technique has serious limitations in both quantifying and comparing gene expression profiles from non-model animals. RNA-seq, meanwhile, can be applied to reconstruct the complete and high-resolution transcriptomes across all species. To build the transcriptome, several methods based on RNA-seq have been developed, usually falling into two main classes:the ‘genome-guided’ (Guttman et al, 2010; Trapnell et al,2010) and genome-independent classes (De novo assembly) (Birol et al, 2009; Schulz et al, 2012). The first methods rely on a reference genome to initially map all the RNA-seq reads to the genome and then assemble overlapping reads into transcripts. Unfortunately, the genome-guided method is not always effective, both because despite a large drop in the cost of nextgeneration sequencing, the study of a complete genome is still costly and difficult, especially for non-model organisms, and because the particular model being studied may be sufficiently different from its reference genome because it comes from a different strain or line.Consequently, de novo assembly is particularly suitable for application to obtain accurate reconstructions. A recent study reported a large RNA-seq data set obtained from six organs of nine different mammals (human,chimpanzee, bonobo, gorilla, orangutan, macaque,mouse, opossum, and platypus) and one bird (chicken),including both males and females (Brawand et al, 2011),demonstrated the utility of applying comparative systems biology between human and non-model animals and elucidated the large evolutionary gaps among these model organisms.
Determining the expression states (i.e., the presence or absence) of genes with low abundance is a challenge for microarray. Consequently, the reconstruction of the actual biological networks (e.g., protein-protein interaction, transcriptional regulation network, or metabolic network) in either human or animal models in a specific condition is very difficult, not to say anything of the difficulty in comparing the dynamic networks(Farmer et al, 2012). Moreover, abnormal variations in alternative splicing are also implicated in disease, thus alternative splicing is a critical factor to consider in building a proper and viable animal model (Luco et al,2011). Unfortunately, obtain the precise alternative splicing map using the microarray technique is almost impossible.
RNA-seq data is highly replicable with relatively little technical variation. For many purposes, RNA-seq may be sufficient to sequence each mRNA sample once.The information obtained in a single lane of RNA-seq data appears to be comparable to that in a single array,and is therefore useful in enabling the identification of differentially expressed genes and allowing for additional,further analyses, such as detection of low-expressed genes, novel transcripts and alternative splice variants. In using this method, researchers can obtain actual biological networks in both human and animal models,and garner biologically meaningful results by comparing between these two networks. Rowley et al (2011), for example, compared the actual transcriptome in platelets between humans and mice, providing critical information used in the design of mouse models of hemostasis and in catalyzing the discovery of new platelet functions..
ChIP-seq for detecting regulation changes
Molecular interactions between proteins and DNA play an essential role in the regulation of gene expression(Cawley et al, 2004; Pokholok et al, 2006). Accordingly,changes in protein–DNA interactions between human and animal models may lead to the divergent functions of homologous pathways (Brown et al, 2011; Greber et al,2010), which is also an important aspect of comparative systems biology.
Chromatin immunoprecipitation (ChIP) followed by genomic tiling microarray hybridization (ChIP-chip) has become the most widely used approach for genome-wide identification and characterization of in vivo protein-DNA interactions during the past decade (Ho et al, 2011).Specifically, when applied to the study of animal models of human disease, CHIP-chip approaches led to many important discoveries in relation to transcriptional regulation (Chen et al, 2008), epigenetic regulation through histone modification (Heintzman et al, 2007),and evolution of protein-DNA interactions (Kim et al,2007).
Like the microarray technique, CHIP-chip also has some limitations arising from the innate characteristics of microarray hybridization. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) makes it possible to obtain the accurate information about the genome-wide profiling of DNA- protein interaction. Compared to the CHIP-chip, ChIP-seq has a higher resolution, fewer artifacts, a larger coverage and a more extensive dynamic range (Blow et al, 2010; Johnson et al, 2007; Mardis 2007; Schmid & Bucher, 2007; Visel et al, 2009).Subsequently, we will introduce the practical applications of ChIP-seq in comparison between human and animals, including (1) identifying the regulatory sequences, and (2) tracing the evolution of epigenetic regulation.
The human genome project, while obtaining the complete genomic sequences, leaves open the question of how to identify the regulatory sequences that control the spatial and temporal expression of genes unanswered(Birney et al, 2007; McGaughey et al, 2008). Through applying the ChIP-seq techniques with the enhancerassociated protein p300 from mouse embryonic heart tissue, Blow et al (2010) made an attempt to identify candidate heart enhancers on genomic scale, revealing that most of the candidate heart enhancers were less deeply conserved in vertebrate evolution when compared to the enhancers that are active in other tissues. Such methods could also be applied to identification of other transcriptional factors (TFs), and therefore are helpful in the reconstruction of the transcriptional regulation network in human and animal models. Thankfully, the decreasing cost of ChIP-seq has extended the comparative systems biology investigation to some TFs.For example, Schmidt et al (2010) used ChIP-seq to determine experimentally the genome-wide occupancy of two TFs, i.e., CCAAT/enhancer-binding protein alpha and hepatocyte nuclear factor 4 alpha, in the livers of five vertebrates, revealing large interspecies differences in transcriptional regulation and providing insight into the evolution of regulatory networks.
Epigenetic regulation is now accepted as being closely associated with human development, and subsequently many developmental disorders may be caused by the dysfunction of this regulation (Gottesman& Hanson, 2005). However, due to the deficient knowledge of this phenomena in other animals, build proper animal models for these studies is difficult.Nevertheless, a recent study that employed the CHIP-seq technique to investigate the epigenetic regulation of histone H3 K4 on frogs (Xenopus tropicalis), revealed a hierarchy in the spatial control of zygotic gene activation(Akkers et al, 2009). Taken together, these advances lead us to speculate that the applications of CHIP-seq in comparative systems biology will be of great help in understanding embryonic diseases.
Despite the advances that ChIP-seq offers,researchers should be cautious when performing ChIP-seq analysis because the experimental steps in ChIP-seq involve several potential sources of artefacts (Park,2009). For example, one challenge in this technique is that the identified enriched regions are of different types for different proteins (for details, refer to (Park, 2009)).The other potential source of artefacts comes from the divergence of both protein and DNA; therefore when using this analysis, the control experiment should be designed carefully.
Perspective applications of comparative systems biology
Comparative systems biology takes advantage of the systematic information from other organisms and can be used to great effect in studying human physiology and disease. Over the coming years, we expect many exciting developments as this field evolves in several potential directions.
Dynamic networks
Biological systems exhibit complex dynamic behavior, enabling cells to react to various conditions or cell states such as cell cycle progression (Zhu et al, 2007).Although static biological systems have been well studied (Benfey & Mitchell-Olds, 2008; Gianchandani et al, 2006; Macilwain, 2011; Werner, 2007), the information gained from such studies is of limited use in moving forward due to the fact that the static interactions are often identified from cells exposed to a single condition or at a single time point, i.e., under nonnative conditions. Only recently have approaches emerged that attempt to analyze the dynamics of complex biological networks. For transcriptional regulatory interactions,ChIP-seq technology is likely to become increasingly popular as it can be used to uncover contextual and temporal variation. For context-specific metabolic network, RNA-seq could provide the dynamic states of metabolic enzymes.
Biological engineering
The ability to manipulate living organisms is at the heart of a range of emerging technologies aimed at addressing critical problems in environment, energy, and health. Because of their complexity and interconnectivity, however, animal models have been less than useful for engineered manipulation. To move forward with employing animal models with greater breadth and application, we vitally need more detailed information that can be obtained using new methods like those outlined in the present study. for instance utilizing real-time RNA-seq technique to obtain the information about the effects of perturbations on biological systems(Faith et al, 2011). Next-generation sequencing technology and the concurrent development of applications for it are a fast-moving area of biomedical research that greatly advance the development of comparative systems biology.
Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM,Francoijs KJ, Stunnenberg HG, Veenstra GJC. 2009. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus Embryos. Dev Cell, 17(3): 425-434.
Alini M, Eisenstein SM, Ito K, Little C, Kettler AA, Masuda K,Melrose J, Ralphs J, Stokes I, Wilke HJ. 2008. Are animal models useful for studying human disc disorders/degeneration? Eur Spine J,17(1): 2-19.
Alvarado AS, Tsonis PA. 2006. Bridging the regeneration gap: genetic insights from diverse animal models. Nat Rev Genet, 7(11): 873-884.
Barabasi AL, Gulbahce N, Loscalzo J. 2011. Network medicine: a network-based approach to human disease. Nate Rev Genet, 12(1): 56-68.
Benfey PN, Mitchell-Olds T. 2008. From genotype to phenotype:Systems biology meets natural variation. Science, 320(5875): 495-497.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR,Margulies EH, Weng ZP, Snyder M, Dermitzakis ET,Stamatoyannopoulos JA and others. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature, 447(7146): 799-816.
Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao YJ, Hirst M, Schein JE, Horsman DE, Connors JM,Gascoyne RD, Marra MA, Jones SJM. 2009. De novo transcriptome assembly with ABySS. Bioinformatics, 25(21): 2872-2877.
Blow MJ, McCulley DJ, Li ZR, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F and others. 2010. ChIP-Seq
identification of weakly conserved heart enhancers. Nat Genet, 42(9):806-810.
Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P,Weier M, Liechti A, Aximu-Petri A, Kircher M and others. 2011. The evolution of gene expression levels in mammalian organs. Nature,478(7369): 343-348.
Brown S, Teo A, Pauklin S, Hannan N, Cho CHH, Lim B, Vardy L,Dunn NR, Trotter M, Pedersen R and others. 2011. Activin/nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells, 29(8):1176-1185.
Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D,Piccolboni A, Sementchenko V, Cheng J, Williams AJ and others. 2004.Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 116(4): 499-509.
Chakraborty C, Hsu CH, Wen ZH, Lin CS, Agoramoorthy G. 2009.Zebrafish: A complete animal model for in vivo drug discovery and development. Curr Drug Metab, 10(2): 116-124.
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL,Zhang WW, Jiang JM and others. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell, 133(6): 1106-1117.
Cox B, Kotlyar M, Evangelou AI, Ignatchenko V, Ignatchenko A,Whiteley K, Jurisica I, Adamson SL, Rossant J, Kislinger T. 2009.Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol Syst Biol, 5: 279.
Enard W, Khaitovich P, Klose J, Zollner S, Heissig F, Giavalisco P,Nieselt-Struwe K, Muchmore E, Varki A, Ravid R and others. 2002.Intra-and interspecific variation in primate gene expression patterns.Science, 296(5566): 340-343.
Faith JJ, McNulty NP, Rey FE, Gordon JI. 2011. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science,333(6038): 101-104.
Farmer MA, Baliki MN, Apkarian AV. 2012. A dynamic network perspective of chronic pain. Neurosci Lett, 520(2): 197-203.
Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. 2011. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer, 11(2): 135-141.
Friese MA, Montalban X, Willcox N, Bell JI, Martin R, Fugger L. 2006.The value of animal models for drug development in multiple sclerosis.Brain, 129(8): 1940-1952.
Fuentes R, Petersson P, Siesser WB, Caron MG, Nicolelis MAL. 2009.Spinal cord stimulation restores locomotion in animal models of Parkinson's disease. Science, 323(5921): 1578-1582.
Garber M, Grabherr MG, Guttman M, Trapnell C. 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods, 8(6): 469-477.
Gianchandani EP, Brautigan DL, Papin JA. 2006. Systems analyses characterize integrated functions of biochemical networks. Trends Biochem Sci, 31(5): 284-291.
Gottesman II, Hanson DR. 2005. Human development: Biological and genetic processes. Annu Rev Psychol, 56(1): 263-286.
Götz J, Ittner LM. 2008. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci, 9(7): 532-544.
Greber B, Wu GM, Bernemann C, Joo JY, Han DW, Ko K, Tapia N,Sabour D, Sterneckert J, Tesar P and others. 2010. Conserved and divergent roles of FGF signaling in mouse epiblast stem cells and human embryonic stem cells. Cell Stem Cell, 6(3): 215-226.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X,Fan L, Koziol MJ, Gnirke A, Nusbaum C and others. 2010. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol, 28(5):503-510.
Hasenfuss G. 1998. Animal models of human cardiovascular disease,heart failure and hypertrophy. Cardiovasc Res, 39(1): 60-76.
Heintzman ND, Stuart RK, Hon G, Fu YT, Ching CW, Hawkins RD,Barrera LO, Van Calcar S, Qu CX, Ching KA and others. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet, 39(3): 311-318.
Henkelman RM. 2010. Systems biology through mouse imaging centers: experience and new directions. Annu Rev Biomed Eng, 12(1):143-166.
Ho JWK, Bishop E, Karchenko PV, Negre N, White KP, Park PJ. 2011.ChIP-chip versus ChIP-seq: Lessons for experimental design and data analysis. Bmc Genomics, 12: 134.
Huh Y, Ju MS, Park H, Han SJ, Bang YM, Ferris CF, Koppe GA, King JA, Kim ML, Kim DJ and others. 2010. Clavulanic acid protects neurons in pharmacological models of neurodegenerative diseases.Drug Develop Res, 71(6): 351-357.
Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E,Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A and others. 2004.Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature, 431(7011): 946-957.
Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science, 316(5830):1497-1502.
Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome.Cell, 128(6): 1231-1245.
Liao BY, Zhang JZ. 2006. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol Biol Evol,23(3): 530-540.
Liao BY, Zhang JZ. 2008. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci USA, 105(19): 6987-6992.
Lieschke GJ, Currie PD. 2007. Animal models of human disease:zebrafish swim into view. Nat Rev Genet, 8(5): 353-367.
Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. 2011.Epigenetics in alternative Pre-mRNA splicing. Cell, 144(1): 16-26.
Macilwain C. 2011. Systems biology: evolving into the mainstream.Cell, 144(6): 839-841.
Mardis ER. 2007. ChIP-seq: welcome to the new frontier. Nat Methods,4(8): 613-614.
Marques AC, Vinckenbosh N, Brawand D, Kaessmann H. 2008.Functional diversification of duplicate genes through subcellular adaptation of encoded proteins. Genome Biol, 9(3): 5R4.
McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA,McCallion AS. 2008. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res,18(2): 252-260.
Merchenthaler I, Shughrue PJ. 2005. Neuroprotection by estrogen in animal models of ischemia and Parkinson's disease. Drug Develop Res,66(2): 172-181.
Miller JA, Horvath S, Geschwind DH. 2010. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways.Proc Natl Acad Sci USA, 107(28): 12698-12703.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008.Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 5(7): 621-628.
Nestler EJ, Hyman SE. 2010. Animal models of neuropsychiatric disorders. Nat Neurosci, 13(10): 1161-1169.
Northoff G. 2009. Comparison between animal models and human imaging findings in major depressive disorder-convergences and divergences. Biol Psychiat, 65(8): 18S-18S.
Ogawa Y, Arakawa K, Kaizu K, Miyoshi F, Nakayama Y, Tomita M.2008. Comparative study of circadian oscillatory network models of Drosophila. Artif Life, 14(1): 29-48.
Park PJ. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet, 10(10): 669-680.
Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA.2006. Activated signal transduction kinases frequently occupy target genes. Science, 313(5786): 533-536.
Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. 2011. Genome-wide RNA-seq
analysis of human and mouse platelet transcriptomes. Blood, 118(14):E101-E111.
Schmid CD, Bucher P. 2007. ChIP-Seq data reveal nucleosome architecture of human promoters. Cell, 131(5): 831-832.
Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD,Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S and others. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science, 328(5981): 1036-1040.
Schulz MH, Zerbino DR, Vingron M, Birney E. 2012. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels.Bioinformatics, 28(8): 1086-1092.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol, 28(5): 511-515.
van den Heuvel S, Dyson NJ. 2008. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Bio, 9(9): 713-724.
Visel A, Blow MJ, Li ZR, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F and others. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature, 457(7231): 854-858.
Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 10(1): 57-63.
Werner E. 2007. All systems go. Nature, 446(7135): 493-494.
Yanai I, Graur D, Ophir R. 2004. Incongruent expression profiles between human and mouse orthologous genes suggest widespread neutral evolution of transcription control. Omics, 8(1): 15-24.
Zhao YQ, Sheng ZZ, Huang JF. 2012. A systematic analysis of heart transcriptome highlights divergent cardiovascular disease pathways between animal models and humans. Mol Biosyst, 8(2): 504-510.
Zhu XW, Gerstein M, Snyder M. 2007. Getting connected: analysis and principles of biological networks. Gene Dev, 21(9): 1010-1024.
