肿瘤坏死因子α在肺炎支原体感染小鼠主要脏器和血清中的表达及其相关机制
2013-09-19宋星宇车广华胡起波
宋星宇,车广华,高 航,胡起波,许 忠,潘 薇,徐 坤
(1.吉林大学第二医院儿科,吉林 长春 130041;2.吉林大学白求恩医学院病理学系,吉林 长春 130021;3.吉林大学公共卫生学院营养与食品卫生学教研室,吉林 长春 130021)
导致Na+-K+泵失活,还可诱导其他炎性介质的释放,导致其他细胞损伤。TNF-α不仅在原位引起肺组织损伤,还可进入血液循环,与其他细胞因子协同作用进一步加重全身炎症反应[2],导致肺外组织的损伤。
1 材料与方法
1.1 动物和主要试剂 选择18~20g雌性清洁级
BALB/C小鼠72只,购于吉林大学白求恩医学院实验动物中心,动物合格证号:SYXK(吉)2008-001C/0011;PPLO肉汤培养基购于青岛海博生物技术有限公司,MP菌液购于上海宝米科生物科技有限公司,阿奇霉素购于辉瑞制药有限公司,小鼠TNF-αELISA试剂盒购于上海蓝基生物公司,SP免疫组织化学试剂盒(KIT-9710)和二氨基联苯胺(diaminobenzidine,DAB)显色剂购于福州迈新生物技术开发有限公司,鼠抗TNF-α多克隆抗体购于北京博奥森生物技术有限公司,Trizol购于Invitrogen公司,实验用引物、DL2000(DNA Marker)购于大连宝生物公司,2×Taq PCR MasterMix购于北京天根生物技术有限公司,焦碳酸二乙酯(diethyl pyrocarbonate,DEPC)购于北京鼎国公司,溴化乙锭(EB)购于北京杰美公司,磷酸盐缓冲液(PBS)和10%中性甲醛为本课题组自行配制。
1.2 MP的培养 PPLO肉汤培养基100mL加入小牛血清20mL混匀,培养基中加入100μL MP菌液放于4℃保存。盖硬胶塞并用封口膜封口,于37℃恒温箱中静置培养。每日观察培养基的颜色变化。用11支无菌小试管,编号1~10,取传代培养7d的菌种加入1号试管,余下依次按1∶10递减稀释,11号管作为阴性对照管不加MP菌液,于37℃恒温箱中静置培养,每日观察菌液颜色的变化和液体有无浑浊改变。菌液发生颜色改变的最高稀释度为颜色改变单位。静置培养1个月后,各试管中的培养液颜色改变在⑧号管(10-7)处停止,表明其变色单位(color change unit,CCU)值为10-7,即被检测菌液稀释到10-7时仍有 MP生长,每支试管中液体均无沉淀或浑浊。
1.3 动物模型的建立 72只小鼠随机分为对照组、MP感染组和抗生素治疗组。对照组小鼠第0天接种无菌0.9%生理盐水各50μL;第5、8和9天接种无菌0.9%生理盐水各100μL。MP感染组、抗生素治疗组小鼠第0天接种含1×10-7CCU·mL-1MP液各50μL;第5、8和9天接种含1×10-7CCU·mL-1MP菌液各100μL。治疗组小鼠于第11天起皮下注射阿奇霉素10mg·kg-1(0.2g 溶 于 5% 葡 萄 糖 注 射 液0.1mL,即0.05mL·g-1),连续5d,观察各组小鼠表现并判定MP感染模型建立是否成功。给药结束后,对照组小鼠全部存活、进食、饮水、活动度无明显改变;MP感染组小鼠全部存活,精神状态差,竖毛寒战、咳嗽、口鼻分泌物增多,进食及活动少,大便稀;抗生素治疗组小鼠经治疗后全部存活,精神状态逐渐改善、竖毛、咳嗽减轻或消失、进食及活动度恢复。将小鼠引颈处死,其中一部分肺组织用来提取总RNA及RT-PCR检测,另一部分肺组织与心、肝、肾组织进行组织病理形态学及免疫组织化学染色。
1.4 ELISA法测定小鼠血清中TNF-α表达水平
应用ELISA法检测3组小鼠血清中TNF-α的表达水平,检测方法按照试剂盒说明书进行。
1.5 RT-PCR 选取正常对照组、MP感染组和抗生素治疗组小鼠一部分肺组织,Trizol法提取小鼠肺组织总RNA作为模板,逆转录形成cDNA,取相同的模板进行PCR。PCR反应条件:94℃ 预变性3min,94℃变性30s,58℃、30s,72℃退火1min,35个循环;58℃延伸10min。将产物应用0.8%的琼脂糖凝胶进行电泳,以负极向正极80~100V恒压进行电泳。采用电泳凝胶图像分析系统进行拍照备用,分析目的基因灰度值与GAPDH灰度值的比值。RT-PCR反应条件见表1。
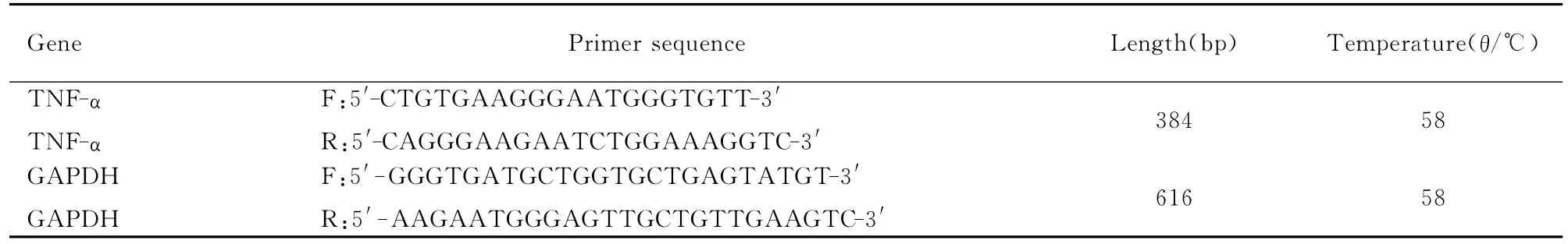
表1 RT-PCR反应条件Tab.1 Test conditions of RT-PCR
1.6 免疫组织化学法检测小鼠各脏器病变程度
取小鼠另一部分肺脏、心脏、肝脏和肾脏石蜡切片,经过二甲苯脱蜡、梯度乙醇水化后用于形态学观察。分别通过切片加热进行抗原修复,采用3%H2O2阻断内源性过氧化物酶活性后,再用PBS进行清洗3次,加与二抗同来源的非免疫动物血清,室温孵育30min后,除去血清,滴加鼠抗TNF-α多克隆抗体。4℃过夜后用PBS清洗切片,加生物素标记的二抗,室温孵育20min后用PBS清洗,加链霉菌抗生物素蛋白-过氧化物酶溶液,室温孵育20min后采用PBS清洗,加新鲜配制的DAB溶液,显微镜下观察3min,自来水冲洗,苏木精复染,梯度酒精脱水,二甲苯透明,中性树脂封片。TNF-α在肺组织中的表达定位于支气管和细支气管上皮细胞的细胞浆和细胞核;TNF-α在肝脏、心脏及肾脏定位于细胞核。以细胞浆和/或细胞核呈棕黄色为阳性表达。400倍视野下选取3~5个阳性视野计数,综合染色强度和阳性率进行评分。染色强度评分标准:阴性0分,染色弱但明显强于阴性对照者为1分,染色清晰者为2分,染色强者为3分。阳性率评分标准:无阳性细胞或阳性率<10%为阴性(-),计0分;阳性率<30%为阳性(+),计1分;31%~60%为2分;>60%为3分。上述2种评分相加≥3分定为阳性。
1.7 统计学分析 采用SPSS 13.0统计软件进行数据处理。各组小鼠血清中TNF-α水平以±s表示,组间比较采用单因素方差分析。
2 结 果
2.1 MP菌株培养情况 于37℃恒温箱内静置培养1个月后,各试管中的培养液颜色改变在⑧号管(10-7)处停止,表明其CCU值为10-7,即被检测菌液稀释到10-7仍有MP生长,每支试管中液体均无沉淀或浑浊。
2.2 小鼠接种情况 小鼠接种后全部存活,对照组小鼠进食、饮水和活动度等无明显改变,状态良好。MP感染组和抗生素治疗组小鼠第1天精神状态略差,活动度降低,进食和排便减少,未见咳嗽等呼吸系统症状;第3天精神状态有所好转,鼻部出现少许分泌物;第6天精神状态较差,活动度降低,进食减少,大便略稀,鼻部分泌物基本同前,并出现咳嗽、竖毛和寒战等症状;于第8和9天接种时乙醚麻醉后小鼠口鼻分泌物明显增加,进食与活动度明显低于对照组。抗生素治疗组小鼠经阿奇霉素治疗后精神状态逐渐好转,咳嗽、竖毛等症状逐渐减轻、消失。进食、活动度逐渐恢复。
2.3 各组小鼠肺和肺外组织病理形态学 病理切片显示,对照组小鼠肺组织和其他脏器无明显炎症反应(图1A,见插页四);MP感染组小鼠肺组织多呈中度或重度炎症改变(图1B,见插页四);抗生素治疗组小鼠的肺组织多呈轻、中度炎症改变(图1C和D,见插页四)。与对照组比较,MP感染组和抗生素治疗组小鼠可见轻度心肌受累,但均无明确感染灶存在(图2A和B,见插页四)。MP感染组和抗生素治疗组各有9只小鼠肝脏呈轻度炎症改变,可见胞浆疏松,但未见感染灶(图2C和D,见插页四),MP感染组和抗生素治疗组共有6只小鼠肾脏受到累及,未见明确感染灶(图2E和F,见插页四)。
2.4 各组小鼠血清中TNF-α水平 MP感染组(0.915±0.015)和抗生素治疗组(0.726±0.029)小鼠血清中TNF-α水平均高于对照组(0.528±0.031)(P<0.01),MP感染组小鼠血清中TNF-α水平高于抗生素治疗组(P<0.01)。
2.5 各组小鼠肺组织TNF-α的表达情况 24只对照组小鼠肺组织中,21只TNF-α表达呈阴性(图3A,见插页四),3只小鼠TNF-α于血管周围表达呈阳性(图3B,见插页四),TNF-α表达阳性率为12.5%;24只MP感染组小鼠中,21只肺组织TNF-α表达呈阳性(图3C和D,见插页四),3只小鼠TNF-α表达呈阴性,TNF-α表达阳性率为87.5%;24只抗生素治疗组小鼠中,9只小鼠肺组织TNF-α表达呈阴性,15只小鼠TNF-α表达呈阳性(中度14只,重度1只)(图3E和F,见插页四),TNF-α表达阳性率为62.5%。MP感染组小鼠肺组织中TNF-α阳性表达率高于对照组和抗生素治疗组(P<0.05);MP感染组和抗生素治疗组可见小鼠心肌TNF-α表达呈阳性(图4A和B,见插页四),但未见感染灶存在,说明心肌有一定程度的受累;MP感染组和抗生素治疗组小鼠中各有6只小鼠肝脏TNF-α表达呈阳性,但未见明确感染灶(图4C和D,见插页四),MP感染组中6只小鼠肾脏TNF-α表达呈阳性(图4E和F,见插页四)。
2.6 各组小鼠肺组织中TNF-αmRNA的表达水平 对照组3只小鼠肺组织中扩增出目的片段,TNF-αmRNA阳性表达率为12.5%。MP感染组21只小鼠肺组织中扩增出目的基因条带,TNF-α mRNA阳性表达率为87.5%。抗生素治疗组15只小鼠肺组织中扩增出目的基因条带,阳性表达率为62.5%。TNF-αmRNA在对照组、MP感染组和抗生素治疗组小鼠肺组织的表达水平分别为0.512±0.095、1.184±0.287和0.889±0.0.213,MP感染组小鼠肺组织中TNF-αmRNA表达水平高于对照组和抗生素治疗组(P<0.05),抗生素治疗组小鼠肺组织中TNF-αmRNA表达水平高于对照组(P<0.05)。见图5。
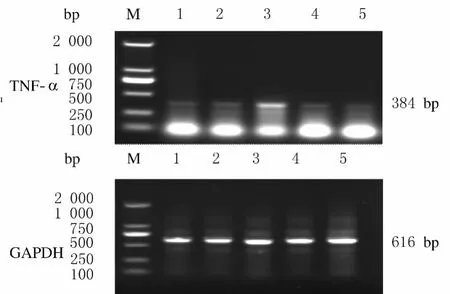
图5 各组小鼠肺组织中TNF-αmRNA表达水平电泳图Fig.5 Electrophoregram of expression levels of TNF-α mRNA in lung tissue of mice in various groups
3 讨 论
MP是已知的能在自然界中独立存活的最小的微生物,含有DNA和RNA,无细胞壁结构,随着研究的深入,人们对MP的认识也不断加深。本实验成功培养了MP,成功建立了MP感染的小鼠模型,并应用ELISA法、免疫组织化学技术和RT-PCR技术对小鼠的血清及组织器官中TNF-α水平进行检测,进一步加深了对MP的认识。MP的基因组非常小,生物合成能力有限,对培养的要求特别高,需在特定的环境中,在富含琼脂和胆固醇的培养基中生长,且培养时间长,具有固定的繁殖周期,以二分裂的方式在体内繁殖[3]。虽然常用的培养基有多种,但本实验选用PPLO肉汤培养基培养MP菌株,除PPLO培养基本身外,还添加了20%的动物血清,为MP的生长提供蛋白质、胆固醇和脂肪酸,10%的新鲜酵母浸液提供核苷酸前体、刺激生长因子和维生素等多种营养物质。MP的最适生长pH值是7.6~7.8,故培养过程中添加0.002%酚红作为指示剂,观察指示剂颜色变化,确保MP生长最佳状态。培养中期,当培养瓶内出现白色絮状物质时,对培养液进行检测,高倍镜下可见大量蓝染小点状或杆状不规则的MP,未见杂菌生长[4]。
可用于MP感染造模的动物模型有很多种,如黑猩猩、棉鼠、地鼠、豚鼠、大鼠和小鼠等。但有些动物不但价格昂贵、饲养环境和感染装置均要求高,而且免疫学检测试剂缺乏,故未采用。本实验采用清洁级BALB/C小鼠作为造模对象[5],不仅因为该小鼠对MP敏感,容易购买,遗传背景清晰,有大量的免疫试剂可供选择。还因该类鼠的基因结构与人的TNF-α的基因结构非常相似,约有80%的同源性。实验采用小剂量、多次接种的方式,实验过程中无一例动物死亡。对照组小鼠无变化或变化不明显,MP感染组和抗生素治疗组小鼠存在呼吸系统症状。抗生素治疗组小鼠应用阿奇霉素连续治疗5d[6]后,小鼠咳嗽症状减轻或消失,无流涕,进食和活动正常,精神状态恢复。MP感染组和抗生素治疗组小鼠肺组织病理切片显示出不同程度的炎症改变,MP感染组重于抗生素治疗组,说明治疗有效。
MP感染的发病机制主要有直接侵入学说、免疫学发病机制和呼吸道上皮粘附学说。在MP感染中,TNF-α作为重要的炎症介质,参与疾病的发生发展。TNF-α是一种具有广泛生物学活性的基因多显性的多肽调节因子,由单核巨噬细胞系统产生,另外中性粒细胞、星状细胞、内皮细胞和平滑肌细胞亦可产生TNF-α[7]。正常情况下,血中存在低水平TNF-α,其不仅是一种有效的肿瘤杀伤因子,也参与机体的免疫防御机能,又在炎症反应、组织损伤、中毒性休克、肿瘤、烧伤和寄生虫病等的病理生理过程中发挥作用[8]。如体内TNF-α水平明显升高可介导炎症的许多病理生理过程,引起局部炎症,介导其他炎症介质的释放,引起组织细胞损伤,导致多器官与系统损害。研究[9]显示血清TNF-α水平越高,全身炎症反应越重。
本实验结果显示:ELISA法和RT-PCR法检测结果一致。为进一步证实实验结果的可靠性,应用免疫组织化学技术[10]从更直观的角度了解病变严重程度,结果显示三种实验结果表达一致。上述结果均证实TNF-α在MP感染的发生发展中起重要作用。
此外,从免疫组织化学结果中可看出,尽管无明确感染灶存在,但TNF-α不仅在MP感染的小鼠肺组织中有阳性表达,在小鼠肝脏、心肌和肾脏组织中同样存在阳性表达,主要考虑存在两种可能:第一种可能原因为MP与机体许多组织、器官存在共同抗原,当MP感染后,可诱导巨噬细胞释放TNF-α,TNF-α亦可反作用于巨噬细胞吞噬抗原,刺激B细胞产生大量与组织器官同源的MP抗体,形成免疫复合物,经血液循环转移到各组织和器官,导致各器官、系统的损伤[11];另外一种可能原因考虑为MP感染可引起免疫系统的变化[12],通常机体存在细胞免疫应答和体液免疫应答,Th1细胞介导细胞免疫应答,以分泌干扰素γ(interferon-γ,IFN-γ)、 白 细 胞 介 素 2(interleukin-2,IL-2)、IL-12和 TNF-α等促炎症介质为特征,可以增强杀伤炎症细胞的细胞毒性作用。Th2细胞介导体液免疫应答,分泌IL-4、IL-5、IL-6和IL-10等细胞因子,以抗炎症介质为主,促进抗体的产生。正常情况下,Thl/Th2通过分泌细胞因子,彼此调节,相互制约,相对平衡,以维持正常的免疫状态。MP感染机体后,细胞免疫占优势,Thl/Th2失衡,导致细胞因子出现紊乱,TNF-α等细胞因子分泌水平增加。TNF-α能诱导中性粒细胞趋化和局部浸润作用,启动炎症反应,增加微血管壁的通透性,激活中性粒细胞及内皮细胞表面黏附受体,引起组织细胞损伤;激活中性粒细胞杀死微生物;刺激单核细胞使其释放血小板源性因子、白三烯和前列腺素等炎性介质,并促进炎性细胞因子包括IL-1、IL-6、TNF-α及IL-8等释放;造成内皮细胞的损害,促进微血栓形成;降低肌细胞跨膜电位,Na+-K+泵失效,并与IL-l协同,加重感染反应[13]。
综上所述,MP感染可引起TNF-α的释放,参与MP感染的病理生理过程,导致机体损害。TNF-α可以作为判断 MP感染严重程度的指标,并可作为判断其预后的指标之一。
[1]Jacobs E.Mycoplasma pneumoniae:now in the focus of clinicians and epidemiologists [J].Eurosurveillance,2012,17(6):1-3.
[2]Lai JF,Zindl CL,et al.Critical role of macrophages and their activation via MyD88-NFkB signaling in lung innate immunity toMycoplasmapneumoniae[J].PLoS One,2010,5(12):1-15.
[3]Dumke R, Jacobs E. Culture-independent multi-locus variable-number tandem-repeat analysis(MLVA) of mycoplasma pneumoniae [J].J Microbiol Methods,2011,86(3):393-396.
[4]Sato C,Manaka S,Nakane D,et al.Rapid imaging of mycoplasma in solution using Atmospheric Scanning Electron Microscopy(ASEM)[J].Biophys Res Commun,2012,417(4):1213-1218.
[5]Kannan TR,Coalson JJ,Cagle M,et al.Synthesis and distribution of CARDS toxin duringMycoplasmapneumoniaeinfection in a murine model [J].J Infect Dis,2011,204(10):1596-1604.
[6]Ríos AM,Fonseca-Aten M,Mejías A,et al.Microbiologic and immunologic evaluation of a single high dose of azithromycin for treatment of experimentalMycoplasma pneumoniaepneumonia[J].AAC,2005,49(9):3970-3973.
[7]Rezende-Oliveira K,Sarmento RR,et al.Production of cytokine and chemokines by human mononuclear cells and whole blood cells after infection withTrypanosomacruzi[J].Rev Soc Bras Med Trop,2012,45(1):45-50.
[8]Persad R, Huynh HQ, Hao L, et al. Angiogenic remodeling in pediatric eosinophilic esophagitis is associated with increased levels of VEGFA,angiogenin,IL-8and activation of the TNF-α-NFκB pathway [J].J Pediatr Gastroenterol Nutr,2012,55(3):251-260.
[9]Kushibiki S.Tumor necrosis factor-α-induced inflammatory responses in cattle [J].Anim Sci J,2011,82(4):504-511.
[10]Nalbantsoy A,Nesil T,Yilmaz-Dilsiz O,et al.Evaluation of the immunomodulatory properties in mice and in vitro antiinflammatory activity of cycloartane type saponins from Astragalus species [J]. J Ethnopharmacol, 2012,139(2):574-581.
[11]Mantawy EM,Tadros MG,Awad AS,et al.Insights antifibrotic mechanism of methyl palmitate:impact on nuclear factor kappa B and proinflammatory cytokines [J].Toxicol Appl Pharmacol,2012,258(1):134-144.
[12]Tubby C,Harrison T,Todd I,et al.Immunological basis offixed airways disease [J].Clin Sci(Lond),2011,121(7):285-296.
[13]Wallis RS.Biologics and infections:lessons from tumor necrosis factor blocking agents [J].Infect Dis Clin North Am,2011,25(4):895-910.
