用于锂离子电池正极材料的分级孔碳/2,5-二巯基-1,3,4-噻二唑/聚噻吩三元复合物
2013-09-17迟婷玉王庚超
迟婷玉 李 涵 王庚超
(华东理工大学材料科学与工程学院,上海市先进聚合物材料重点实验室,超细材料制备与应用教育部重点实验室,上海200237)
1 引言
随着储能电源和电动汽车的迅猛发展,对锂离子电池的性能提出更高要求,开发高能量密度的锂离子电池成为研究重点之一.1−42,5-二巯基-1,3,4-噻二唑(DMcT)作为一种重要的有机多硫化物,因其理论容量高(362 mAh·g−1),作为锂电池正极材料具有潜在的应用价值.5−8但是,DMcT室温下电化学反应速度缓慢,而且放电时产生的小分子硫巯基化合物易溶于有机电解液中,尚不能满足电池的需要.
为了提高DMcT的电化学活性与稳定性,近年来,人们采用导电高分子、9−13纳米碳材料、14,15金属纳米粒子16,17和层状化合物18对其进行复合改性.例如,Oyama等9研究发现,导电聚苯胺对DMcT具有显著的电化学催化作用,且充放电容量接近其理论值.我们研究小组15运用原位聚合法将DMcT负载到磺化石墨烯上,显著提高DMcT的电化学活性,同时循环稳定性也得以改善.Park等17研究证明Pd纳米粒子与DMcT间存在强的相互作用,可形成金属-有机多硫化物络合物,对DMcT的电化学反应也具有催化作用.然而,上述改性方法得到的DMcT复合正极材料的循环性能离实际应用仍有差距.
分级孔碳(HPC)具有比表面积大、电导率高、吸附能力强和稳定性好等特点,19−21有利于活性物质的负载和电解液的扩散,并能提高电荷传输性能,是电极材料的理想载体.近年来,导电聚合物/分级孔碳复合物的制备及其在锂离子电池方面的应用已有报道.22−24但有关分级孔碳负载DMcT复合材料的研究尚未见报道.
本文选用酚醛树脂为碳源,纳米碳酸钙分散液为二次成孔剂,经煅烧和刻蚀制备分级孔碳,并采用KOH对分级孔碳进行活化,制备出活化分级孔碳(aHPC).再以aHPC为基板,通过溶液浸渍法制得aHPC/DMcT复合物,最后运用氧化聚合法将导电聚(3,4-乙烯二氧噻吩)-聚苯乙烯磺酸(PEDOT-PSS)包覆在其表面制备出aHPC/DMcT/PEDOT-PSS复合材料.并系统研究了aHPC和PEDOT-PSS的引入对材料结构形貌和电化学性能的影响.研究发现,PEDOT-PSS导电膜的引入,不仅抑制DMcT在电解液中的溶解性,也对DMcT起到电催化作用,从而提高DMcT的循环稳定性和电化学活性.
2 实验部分
2.1 原材料
3,4-乙烯二氧噻吩(分析纯,Sigma-Aldrich);聚苯乙烯磺酸钠(分析纯,Sigma-Aldrich);过硫酸铵,使用前经重结晶精制处理;2,5-二巯基-1,3,4-噻二唑(分析纯,美国ACROS公司);酚醛树脂(北京富润达有限公司);纳米碳酸钙水分散液(上海华明高技术有限公司,颗粒直径40−60 nm);其它试剂均为分析纯,使用前未经处理.
2.2 活化分级孔碳的制备
分级孔碳是在文献25方法的基础上通过改进而获得,具体步骤如下:将纳米碳酸钙水分散液经过滤、乙醇洗涤后,再经超声处理使纳米碳酸钙均匀分散在乙醇中.将酚醛树脂配成25%的乙醇溶液,再与纳米碳酸钙的乙醇分散液搅拌混合均匀,控制酚醛树脂/纳米碳酸钙的质量比为40:60.然后将混合物中的乙醇挥发掉,得到浅黄色粉末.将混合物粉末在氩气氛中于管式炉中进行炭化煅烧处理.煅烧工艺为从室温以2°C·min−1的速率升温到300 °C,再以5 °C·min−1的速率从300 °C升温到700 °C,再以5 °C·min−1的速率从700 °C升温到850 °C,并在300、700、850 °C分别保温90、60、60 min.将煅烧产物中的氧化钙模板用2 mol·L−1的盐酸去除,用去离子水洗至中性,所得产物为分级孔碳.
将HPC与KOH按质量比1:3混合研磨均匀后,以10 °C·min−1的速率升至800 °C,保温 1 h,自然冷却至室温.产物用大量水洗涤,再在沸腾状态下用0.1 mol·L−1HCl洗涤30 min.最后在200 °C下脱水24 h,即得到活化分级孔碳.
2.3 活化分级孔碳负载2,5-二巯基-1,3,4-噻二唑(aHPC/DMcT)的制备
将0.5 g aHPC粉末抽真空0.5 h,并在真空状态下加入DMcT的无水乙醇溶液,搅拌混合1 h后,通过抽滤除去多余的DMcT,再在室温干燥,如此重复两次,最后在70°C下干燥24 h,得到aHPC/DMcT复合物.
2.4 聚(3,4-乙烯二氧噻吩)-聚苯乙烯磺酸(PEDOT-PSS)的制备
称取2.2 g聚苯乙烯磺酸钠(PSS)溶解在100 mL去离子水中,用2 mol·L−1盐酸将溶液pH值调为2.然后加入0.75 g的3,4-乙烯二氧噻吩(EDOT)单体.再滴加溶有1.7 g过硫酸铵(APS)的200 mL水溶液,室温下搅拌反应24 h.产物经透析后得到PEDOTPSS水溶液.
2.5 聚(3,4-乙烯二氧噻吩)-聚苯乙烯磺酸包覆活化分级孔碳负载2,5-二巯基-1,3,4-噻二唑(aHPC/DMcT/PEDOT-PSS)复合材料的制备
称取0.44 g PSS溶解在20 mL去离子水中,用2 mol·L−1盐酸将溶液pH值调为2.然后加入0.15 g的EDOT单体,搅拌均匀后,再加入0.3 g的aHPC/DMcT.最后将溶有0.34 g APS的40 mL水溶液缓慢滴加到上述混合液中,室温下搅拌反应24 h.产物经离心,去离子水洗涤后得aHPC/DMcT/PEDOT-PSS复合材料.
2.6 电极材料的制备与电池组装
将活性电极材料、乙炔黑、粘合剂(聚偏(二)氯乙烯(PVDF))以8:1:1质量的比例混合研磨均匀,刮涂于集流体铝箔上,干燥后裁成直径为13 mm的极片做正极;以锂片做负极;电解质采用1 mol·L−1LiPF6/碳酸乙烯酯(EC)+碳酸二甲酯(DMC)+碳酸甲乙酯(EMC)(1:1:1)溶液;在Ar气氛手套箱中组装成纽扣电池.
2.7 测试与表征
FTIR图谱采用美国Nicolet 5700红外光谱仪测得,KBr压片.采用日本Rigaku D/Max 2550 VB/PC型X射线衍射仪测试XRD图谱.场发射扫描电镜(FESEM)照片在日本HITACHI S-4800仪器上完成.采用JEOL JEM-1400透射电镜对样品的形貌进行观察.热失重(TG)和示差扫描量热(DSC)分析在TGA/SDTA/851E热分析仪上进行,在氮气氛中,升温速率为10 °C·min−1,温度范围为室温至880 °C.氮吸附-脱附曲线在ASAP 2020仪器上温度为77 K下测试,测试前,样品在393 K下经真空处理8 h.
循环伏安测试和电化学交流阻抗测试均采用CHI 660D电化学工作站测定.循环伏安测试的扫描电压范围为1.8−3.8 V,扫描速率为0.1 mV·s−1.交流阻抗测试的扫描频率为0.01−100 kHz,振幅为5 mV.利用LAND CT2001A测试系统进行充放电测试,电流密度为20−50 mA·g−1,充放电电压范围为1.8−3.8 V.
3 结果与讨论
图1为aHPC,DMcT,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS和PEDOT-PSS的FTIR谱图.由图1a看出,aHPC在1086 cm−1处出现吸收峰,对应于C―OH的弯曲振动,26说明活化后aHPC孔道内引入了羟基,从而赋予其较好的润湿性.DMcT在1600−500 cm−1范围内存在诸多吸收峰,分别与噻唑环上N―N,C―N和C―S等基团的振动有关(图1b).对于aHPC/DMcT复合物,也出现了DMcT的特征峰,但强度减弱,这可能是DMcT进入到aHPC孔内缘故(图1c).PEDOT-PSS在1520和1330 cm−1处吸收峰与噻吩环骨架C=C和C―C伸缩振动有关,831 cm−1处吸收峰归属于C―S键的伸缩振动,1141和1087 cm−1处吸收峰则与乙烯二氧基的振动有关,271033和1003 cm−1处吸收峰说明 SO3−基团的存在(图1e).aHPC/DMcT/PEDOT-PSS复合物也出现PEDOT-PSS的吸收峰,而DMcT的吸收峰则不明显(图1d).
图2给出了aHPC,DMcT,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS和PEDOT-PSS的XRD图.我们发现,aHPC在2θ为15°−35°范围内出现了一个宽的衍射峰,说明aHPC为非晶态结构(图2a).DMcT显示多个强的衍射峰,表明其呈现良好的晶态结构(图2b).DMcT与aHPC复合后,DMcT的结晶衍射峰变得非常弱(图2c),这可能由于DMcT进入到aHPC孔内,抑制了其结晶行为,导致DMcT呈现非晶态特征.对于PEDOT-PSS(图2e)而言,在18.1°和25.9°处出现两个弱而宽的衍射峰,这也说明PEDOT-PSS为非晶态结构.aHPC/DMcT/PEDOT-PSS复合物出现PEDOT-PSS和aHPC的宽衍射峰,但未出现DMcT的结晶衍射峰(图2d),这是aHPC/DMcT表面被PEDOT-PSS所覆盖的缘故.
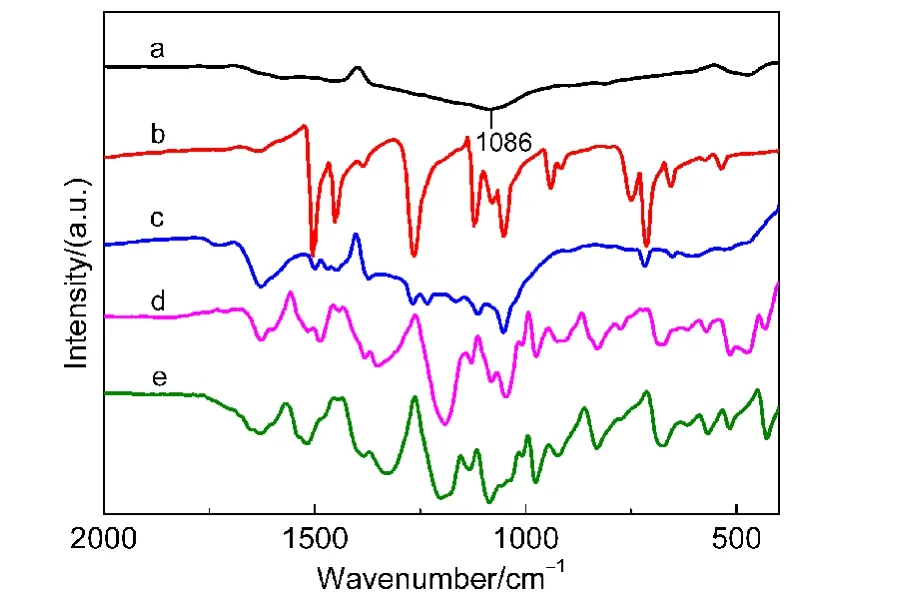
图1 (a)aHPC,(b)DMcT,(c)aHPC/DMcT,(d)aHPC/DMcT/PEDOT-PSS和(e)PEDOT-PSS的FTIR谱图Fig.1 FTIR spectra of(a)aHPC,(b)DMcT,(c)aHPC/DMcT,(d)aHPC/DMcT/PEDOT-PSS,and(e)PEDOT-PSS

图2 (a)aHPC,(b)DMcT,(c)aHPC/DMcT,(d)aHPC/DMcT/PEDOT-PSS和(e)PEDOT-PSS的XRD谱图Fig.2 XRD patterns of(a)aHPC,(b)DMcT,(c)aHPC/PDMcT,(d)aHPC/DMcT/PEDOT-PSS,and(e)PEDOT-PSS
图3 显示了aHPC,aHPC/DMcT和aHPC/DMcT/PEDOT-PSS的FESEM和TEM照片.可以看出,aHPC的形貌表现为分布均匀且相互贯通的多孔结构,孔径尺寸在40−60 nm范围内.与DMcT复合后,aHPC的大多数的孔道被DMcT所充填,aHPC表面几乎看不出片状物,说明几乎所有的DMcT均进入到aHPC孔内.从TEM照片(图3d)也可看出孔内黑色部分为DMcT粉末负载,且负载较均匀,进一步证实DMcT均匀分散在aHPC孔内.其原因是活化后aHPC孔表面含较多―OH,DMcT在孔中的浸润性较好,故能全部地寄生在其孔内.当涂覆PEDOTPSS后,三元复合物的多孔结构很难发现(图3e),TEM照片进一步显示,aHPC/DMcT表面均匀包覆了一层PEDOT-PSS膜(图3f).
图4为aHPC,aHPC/DMcT和aHPC/DMcT/PEDOTPSS的氮气吸附-脱附等温曲线.由图可看出,aHPC的等温曲线为I/II型,在极低压力下表现为微孔吸附,当微孔吸附饱和后,随相对压力的增大,吸附平台趋向于平缓,之后出现很明显的滞后环,为毛细凝聚导致,意味着材料含大量的介孔,经测试其BET比表面积高达666 m2·g−1.对于aHPC/DMcT复合物,其微孔吸附消失,且滞后环明显减小,比表面积降为117 m2·g−1,这是由于DMcT寄生在aHPC孔内,使孔体积和比表面急剧缩小.而aHPC/DMcT/PEDOT-PSS复合物的滞后环进一步减小,比表面积降低到34 m2·g−1,其原因是表面包覆一层PEDOTPSS薄膜后,复合物的大部分孔被封闭,故孔体积和比表面积进一步减小.

图3 (a)aHPC,(c)aHPC/DMcT和(e)aHPC/DMcT/PEDOT-PSS的FESEM照片;(b)aHPC,(d)aHPC/DMcT,和(f)aHPC/DMcT/PEDOT-PSS的TEM照片Fig.3 FESEM images of(a)aHPC,(c)aHPC/DMcT,and(e)aHPC/DMcT/PEDOT-PSS;TEM images of(b)aHPC,(d)aHPC/DMcT,and(f)aHPC/DMcT/PEDOT-PSS
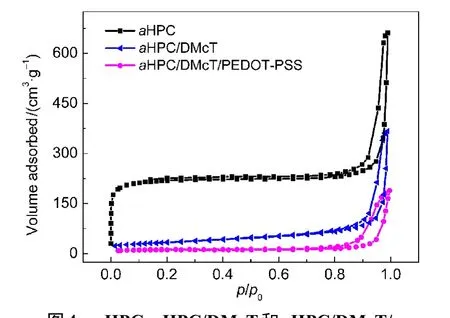
图4 aHPC,aHPC/DMcT和aHPC/DMcT/PEDOT-PSS的氮气吸附-脱附曲线Fig.4 N2adsorption-desorption isotherms of aHPC,aHPC/DMcT,and aHPC/DMcT/PEDOT-PSS
图5(A)给出了DMcT,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS和PEDOT-PSS的热失重曲线.DMcT单体在160°C附近开始分解,分解速度最快时的温度约为230°C,在720°C时分解完毕.而aHPC/DMcT复合物在200°C开始分解,且分解速度要缓于DMcT,其原因可能是DMcT寄生在aHPC孔内部,减缓了DMcT的分解.由aHPC/DMcT在880°C的重量残留率为48%,可推算出DMcT在aHPC内的负载量约为52%.PEDOT-PSS的重量损失主要集中在230−400 °C,这主要归因于PSS的降解;880 °C下重量保持率高达50%,说明PEDOT的热稳定性突出.对于aHPC/DMcT/PEDOT-PSS复合物而言,PEDOT-PSS的包覆使复合物的开始分解温度提高到230°C左右,这表明PEDOT-PSS包覆层的存在对DMcT起着良好的保护作用.DSC曲线显示(图5(B)),DMcT在173°C处出现一个明显的吸热峰,与DMcT的结晶熔融有关.而aHPC/DMcT复合物在173°C附近未出现吸热峰,表明DMcT以非晶态存在,这与XRD的研究结果一致,进一步说明DMcT进入到aHPC孔内.

图5 (A)DMcT,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS和PEDOT-PSS的热失重曲线;(B)DMcT和aHPC/DMcT的差示扫描量热曲线Fig.5(A)Thermogravimetric(TG)curves for DMcT,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS,and PEDOT-PSS;(B)differential scanning calorimetry(DSC)curves for DMcT and aHPC/DMcT
表1为DMcT,aHPC,PEDOT-PSS,aHPC/DMcT,aHPC/DMcT/PEDOT-PSS的电导率.DMcT为电绝缘材料,当与aHPC复合后,aHPC/DMcT的电导率增加到0.61 S·cm−1.PEDOT-PSS的电导率很高,可达12 S·cm−1,显示良好的导电性.PEDOT-PSS包覆aHPC/DMcT后,复合材料的电导率提高到1.4S·cm−1.
图6为DMcT,aHPC/DMcT和aHPC/DMcT/PEDOTPSS的循环伏安曲线.我们发现,DMcT在2.1和2.4 V处出现两个弱还原峰,在3.3 V出现弱氧化峰,其电化学活性较差,并呈现出明显的极化现象(图6a).当DMcT寄生到aHPC孔内形成aHPC/DMcT后(图6b),氧化还原峰的强度明显增强,氧化峰与还原峰的电位差变小,由0.9 V(DMcT)减小到0.7 V(aHPC/DMcT),且极化现象明显改善.这是因为aHPC的纳米孔结构和良好导电性使得DMcT以纳米尺度负载在aHPC孔内部,大大改善DMcT的电荷传输性能,对DMcT的电化学反应有明显促进作用.aHPC/DMcT/PEDOT-PSS也出现两个还原峰(2.5和2.6 V)和一个氧化峰(3.2 V),且峰强度相对aHPC/DMcT进一步增强,氧化峰与还原峰的电位差也进一步减小,仅为0.6 V.其原因可能是PEDOT-PSS的引入,不仅提高了复合材料的导电性,也对DMcT起到电催化作用,故材料的电化学活性更佳.

表1 DMcT,aHPC,PEDOT-PSS,aHPC/DMcT和aHPC/DMcT/PEDOT-PSS的电导率Table 1 Electrical conductivities of DMcT,aHPC,PEDOT-PSS,aHPC/DMcT,and aHPC/DMcT/PEDOT-PSS

图6 (a)DMcT,(b)aHPC/DMcT和(c)aHPC/DMcT/PEDOT-PSS电极在扫描速率为0.1 mV·s−1时的循环伏安曲线Fig.6 Cyclic voltammograms of(a)DMcT,(b)aHPC/DMcT,and(c)aHPC/DMcT/PEDOT-PSS electrodes at a scan rate of 0.1 mV·s−1
图7显示了aHPC/DMcT和aHPC/DMcT/PEDOTPSS复合物的首次充放电曲线.可以看出,两种复合物在2.8和2.6 V附近均出现两个放电平台,可归因于放电过程中产生不同物质,如三聚物、二聚物等.28从aHPC/DMcT的放电曲线看出,其首次放电容量为 236 mAh·g−1(以 DMcT质量计算).对于aHPC/DMcT/PEDOT-PSS复合物而言,其首放容量高达281 mAh·g−1,明显高于二元复合物的容量.这是因为在aHPC/DMcT表面包覆一层导电PEDOTPSS薄膜,不仅提高了材料的导电性能,而且PEDOT-PSS中的噻唑共轭环可以吸附电荷和解吸附电荷,更加利于电荷传输,起着电催化剂的作用,使得DMcT的电化学活性提高.

图7 aHPC/DMcT和aHPC/DMcT/PEDOT-PSS在电位窗口为1.8−3.8 V和电流密度为20 mA·g−1时的首次充放电曲线Fig.7 The first charge-discharge curves of aHPC/DMcT and aHPC/DMcT/PEDOT-PSS electrodes under the potential window of 1.8−3.8 V at a current density of 20 mA·g−1
图8 (A)反映了aHPC/DMcT和aHPC/DMcT/PEDOT-PSS的循环稳定性.aHPC/PDMcT的放电容量在第2次循环后减少到125 mAh·g−1,20次循环后仅为65 mAh·g−1.这是因为在充放电循环过程中,其孔内DMcT活性物质逐渐溶解到电解液中,故其容量衰减得较快.当在其表面包覆PEDOT-PSS薄膜后,aHPC/DMcT/PEDOT-PSS的循环性能显著提高,6次循环后仍能达到150 mAh·g−1,7次循环开始,aHPC/DMcT/PEDOT-PSS复合物的容量衰减变缓,20次充放电后,仍能保持138 mAh·g−1的放电比容量,容量保持率可达49.1%,显示出较好的循环性能.上述结果优于PDMcT-PEDOT/PEDOT-PSS(6次循环后,容量保持率低于20%),11PPy包覆PDMcT(3次循环后,容量保持率60%)10和磺化石墨烯负载PDMcT复合正极材料(10次循环后,容量保持率46.3%).15这归因于aHPC/DMcT表面包覆一层PEDOT-PSS导电薄膜,一定程度上阻止了DMcT溶于电解液中,从而显著改善DMcT的循环性能.然而从前面的电镜照片发现,aHPC/DMcT的表面并不是被PEDOT-PSS薄膜完全覆盖,故容量仍有衰减.图8(B)显示了aHPC/DMcT和aHPC/DMcT/PEDOTPSS在不同电流密度下的倍率性能.可见,与aHPC/DMcT二元复合物相比,包覆PEDOT-PSS后的三元复合物的倍率性能有较好改善,在较大电流密度下的放电容量保持率明显提高,并且显现出良好的容量回复性能.
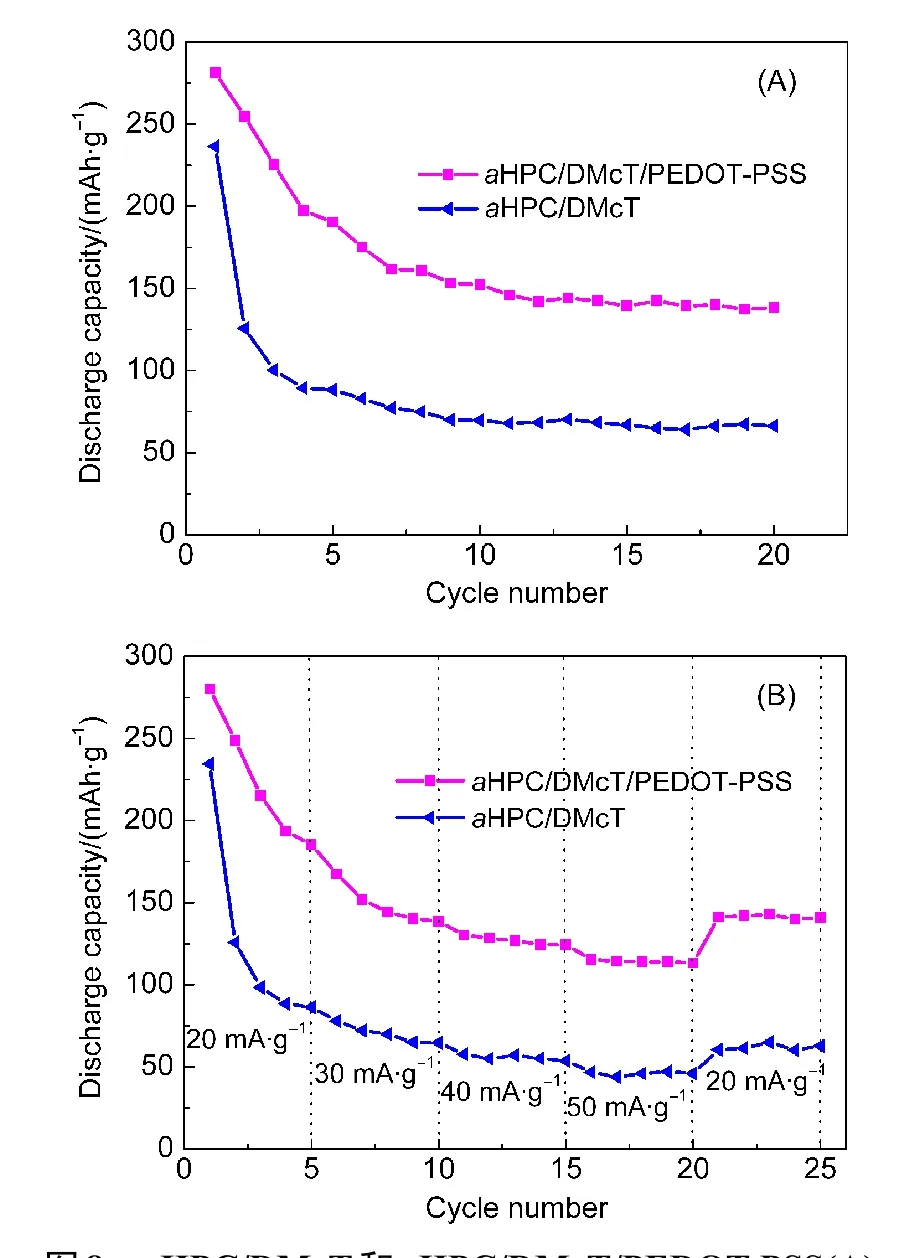
图8 aHPC/DMcT和aHPC/DMcT/PEDOT-PSS(A)在电流密度为20 mA·g−1时的循环性能(A)及其不同电流密度下的倍率性能(B)Fig.8 Cycling performance at a current density of 20 mA·g−1(A)and rate capabilities at various current densities(B)for aHPC/PDMcT and aHPC/DMcT/PEDOT-PSS electrodes
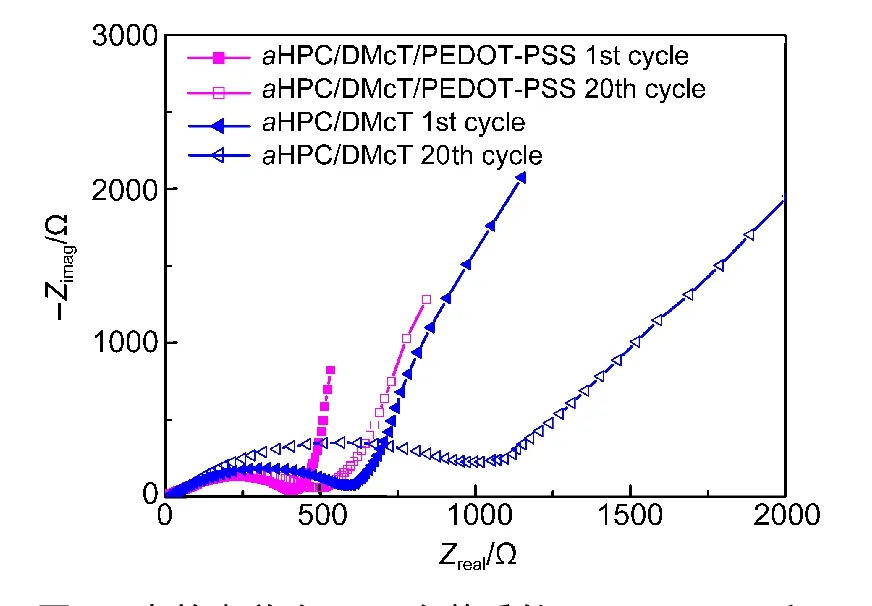
图9 充放电首次及20次前后的aHPC/DMcT和aHPC/DMcT/PEDOT-PSS的交流阻抗图Fig.9 Impedance plots of aHPC/PDMcT and aHPC/DMcT/PEDOT-PSS electrodes after the 1st and 20th cycles
图9 显示了aHPC/DMcT和aHPC/DMcT/PEDOTPSS复合物首次和20次循环后的交流阻抗图谱.在高频区,半圆直径对应于电荷转移电阻(Rct),反映了电极和电解质界面间的电荷传输和迁移能力.在低频区,可看到与实轴呈30°−60°的Warburg曲线,为Li+的扩散电阻.与x轴的倾斜角度越大,说明Li+的扩散电阻越小,电池性能越好.aHPC/DMcT和aHPC/DMcT/PEDOT-PSS复合物的初始Rct分别为600和420 Ω.当循环20圈后,aHPC/DMcT的Rct提高到1040 Ω;而对于aHPC/DMcT/PEDOT-PSS复合物,PEDOT-PSS的引入,不仅提高了复合物的导电性,也促进了DMcT的氧化还原反应,使得20次循环后Rct增幅较小,变为550 Ω.也说明了aHPC/DMcT/PEDOT-PSS复合物具有较好的电化学循环性能.
4 结论
以aHPC为载体,分别采用溶液浸渍和原位聚合法制备出aHPC/DMcT和aHPC/DMcT/PEDOTPSS复合物.研究表明,aHPC拥有均匀的连通孔结构,且比表面积高达666 m2·g−1,利于DMcT的负载,能够使DMcT充分进入到aHPC孔道内,aHPC/DMcT复合物首次放电容量为236 mAh·g−1.aHPC/DMcT复合物表面再包覆一层PEDOT-PSS导电薄膜后,三元复合物的首次放电容量高达281 mAh·g−1,20次循环容量的保持率由包覆前的27.5%提高到包覆后的49.1%.这为改善DMcT的电化学性能提供一种新途径.
(1)Armand,M.;Tarascon,J.M.Nature 2008,451,652.doi:10.1038/451652a
(2) Yin,L.C.;Wang,J.L.;Lin,F.J.;Yang,J.;Nuli,Y.Energy Environ.Sci.2012,5,6966.doi:10.1039/c2ee03495f
(3) Wang,J.Z.;Lu,L.;Choucair,M.;Stride,J.A.;Xu,X.;Liu,H.K.J.Power Sources 2011,196(16),7030.doi:10.1016/j.jpowsour.2010.09.106
(4) Xin,S.;Guo,Y.G.;Wan,L.J.Accounts Chem.Res.2012,45(10),1759.doi:10.1021/ar300094m
(5) Visco,S.J.;DeJonghe,L.C.J.Electrochem.Soc.1988,135(12),2905.doi:10.1149/1.2095460
(6)Yu,L.;Wang,X.H.;Li,J.;Jing,X.B.;Wang,F.S.Chem.J.Chin.Univ.2000,21(2),311.[于 雷,王献红,李 季,景遐斌,王佛松.高等学校化学学报,2000,21(2),311.]
(7) Zhang,J.H.;Zhang,Y.S.;Zheng,M.P.;Qi,L.;Feng,B.;Li,L.Acta Phys.-Chim.Sin.2007,23(Supp),51.[张敬华,张永生,郑绵平,其 鲁,冯 波,李 立.物理化学学报,2007,23(Supp),51.]doi:10.3866/PKU.WHXB2007Supp12
(8) Henderson,J.C.;Kiya,Y.;Hutchison,G.R.;Abruna,H.D.J.Phys.Chem.C 2008,112(10),3989.doi:10.1021/jp076774k
(9) Oyama,N.;Tatsuma,T.;Sato,T.;Sotomura,T.Nature 1995,373,598.doi:10.1038/373598a0
(10) Xue,L.J.;Li,J.X.;Hu,S.Q.;Zhang,M.X.;Zhou,Y.H.;Zhan,C.M.Electrochem.Commun.2003,5(10),903.doi:10.1016/j.elecom.2003.08.018
(11) Kiya,Y.;Hutchison,G.R.;Henderson,J.C.;Sarukawa,T.;Hatozaki,O.;Oyama,N.;Abruña,H.D.Langmuir 2006,22(25),10554.doi:10.1021/la061213q
(12) Kiya,Y.;Iwata,A.;Sarukawa,T.;Henderson,J.C.;Abruña,H.D.J.Power Sources 2007,173,522.doi:10.1016/j.jpowsour.2007.04.086
(13) Chi,T.Y.;Li,H.;Li,X.W.;Bao,H.;Wang,G.C.Electrochim.Acta 2013,96,206.doi:10.1016/j.electacta.2013.02.100
(14) Canobre,S.C.;Almeida,D.A.L.;Fonseca,C.P.;Neves,S.Electrochem.Acta 2009,54(26),6383.doi:10.1016/j.electacta.2009.06.002
(15) Jin,L.F.;Wang,G.C.;Li,X.W.;Li,L.B.J.Appl.Electrochem.2011,41(4),377.doi:10.1007/s10800-010-0246-z
(16) Ortega,P.;Vera,L.;Guzman,M.Macromol.Chem.Phys.1997,198(9),2949.doi:10.1002/macp.1997.021980923
(17) Park,J.E.;Park,S.G.;Koukitu,A.;Hatozaki,O.;Oyama,N.Synth.Met.2004,140(2−3),121.doi:10.1016/j.synthmet.2003.04.001
(18) Wang,G.C.;Jin,L.F.;Ye,J.K.;Li,X.W.Mater.Chem.Phys.2010,122(1),224.doi:10.1016/j.matchemphys.2010.02.038
(19) Xing,W.;Huang,C.C.;Zhuo,S.P.;Yuan,X.;Wang,G.Q.;Hulicova-Jurcakova,D.;Yan,Z.F.;Lu,G.Q.Carbon 2009,47(7),1715.doi:10.1016/j.carbon.2009.02.024
(20) Yi,J.;Li,X.P.;Hu,S.J.;Li,W.S.;Zhou,L.;Xu,M.Q.;Lei,J.F.;Hao,L.S.J.Power Sources 2011,196(16),6670.doi:10.1016/j.jpowsour.2010.12.017
(21) Hasegawa,G.;Kanamori,K.;Nakanishi,K.Microporous Mesoporous Mat.2012,155(1),265.doi:10.1016/j.2012.02.001(22) Yang,J.;Zhou,X.Y.;Zou,Y.L.;Tang,J.J.Electrochim.Acta 2011,56(24),8576.doi:10.1016/j.electacta.2011.07.047
(23) Guo,B.K.;Wang,X.Q.;Fulvio,P.F.;Chi,M.F.;Mahurin,S.M.;Sun,X.G.;Dai,S.Adv.Mater.2011,23(40),4661.doi:10.1002/adma.201102032
(24) Kim,Y.;Jo,C.;Lee,J.;Lee,C.W.;Yoon,S.J.Mater.Chem.2012,22,1453.doi:10.1039/c1jm15053g
(25) Pope,J.M.;Sato,T.;Shoji,E.;Oyama,N.;White,K.C.;Buttry,D.A.J.Electrochem.Soc.2002,149(7),A939.doi:10.1149/1.1482768
(26) Sun,X.M.;Li,Y.D.Angew.Chem.Int.Edit.2004,43(5),597.doi:10.1002/anine.200353212
(27) Han,Y.Q.;Lu,Y.Synth.Met.2008,158(19−20),744.doi:10.1016/j.synthmet.2008.04.028
(28) Li,J.X.;Zhan,H.;Zhou,Y.H.Electrochem.Commun.2003,5(7),555.doi:10.1016/S1388-2481(03)00121-8
