三环已基锡R-扁桃酸酯配合物的合成、结构及性质
2013-09-15张复兴邝代治冯泳兰王剑秋庾江喜蒋伍玖朱小明
张复兴 邝代治 冯泳兰 王剑秋 庾江喜 蒋伍玖 朱小明
(衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳 421008)
有机锡羧酸酯由于具有结构的多变性、丰富的反应性、较强的生物活性和催化活性,多年来一直引起人们的兴趣[1-5]。然而,由于有机锡化合物的高毒性,又使它们的应用受到了一定的限制。相关研究表明,有机锡化合物的生物活性与中心锡原子的构型有关,而中心锡原子的构型决定于直接与锡原子相连的烃基的结构和配体的类型[6-10]。功能化的羧酸配体能极大的改变锡原子的配位方式,显著的影响有机锡羧酸酯的生物活性,从而调节其毒性与生物活性之间的平衡。近年来我们利用功能化的配体合成了一系列具有结构特点的有机锡配合物,并研究了它们的结构和性能[11-18]。为了更进一步揭示有机锡化合物结构与性能的关系,本文合成了手性有机锡三环己基锡R-扁桃酸酯,通过元素分析和红外光谱进行了表征,用X-射线单晶衍射测定了晶体结构,对其结构进行量子化学从头计算,探讨了配合物的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。并研究了配合物的热稳定性、荧光性质和电化性能。
1 实验部分
1.1 试剂与仪器
IRPrestige-21 红 外 光 谱 仪(4 000~400 cm-1,KBr),PE-2400(Ⅱ)元素分析仪,Bruker SMART APEXⅡ单晶衍射仪,TG209F3热分析仪,F-7000型荧光光谱仪,CHI660D电化学工作站,X4数字显微熔点测定仪。所用试剂均为分析纯。
1.2 实验过程
在50 mL圆底烧瓶中,加入 0.770 g(2 mmol)三环己基氢氧化锡、0.304 g(2 mmol)R-扁桃酸、15 mL苯和35 mL乙醇,在电磁搅拌下加热回流分水反应6 h。趁热过滤除去不溶性固体,滤液旋转蒸发除去部分溶剂,放置析出白色固体,用适当的溶剂重结晶得无色透明晶体 0.690 g, 产率 66.51%。 熔点:84~86 ℃。 红外光谱主要吸收峰:3 418.97(m),3 258.87(m),2 920.35(s),2 846.09(m),1 653.01(s),1 601.95(s),1 433.785(m),1 323.22(w),556.49(w),488.98(w)cm-1。元素分析(C26H40O3Sn),计算值(%):C,60.15;H,7.71。实测值(%):C,59.95;H,7.68。
1.3 晶体结构测定
选取尺寸为 0.34 mm×0.23 mm×0.21 mm 单晶体,在Bruker SMART APEXⅡ单晶衍射仪上进行衍射实验,在296(2)K下,用石墨单色化的Mo Kα(λ=0.071073 nm)射线,以 ω-2θ方式扫描收集数据。 在1.60°≤θ≤25.00°范围内共收集 10 501 个衍射点,其中独立衍射点 4580 个(Rint=0.0250),可观察衍射点4 374个(I>2σ(I))。全部数据经Lp校正和吸收校正,以直接法进行晶体结构解析。部分非氢原子坐标随后用差值Fourier合成法确定,理论加氢计算法给出氢原子位置坐标。用SHELX97程序以全矩阵最小二乘法对非氢原子坐标及其各向异性热参数进行修正,残差因子 R1=0.0390,wR2=0.1039。
2 结果与讨论
2.1 晶体结构描述
配合物属斜方晶系,空间群为P212121,晶体学参 数 a=0.806 41(4)nm,b=1.7686 8(9)nm,c=1.834 79(8)nm,V=2.616 9(2)nm3,Z=4,Dc=1.318 g·cm-3,μ(Mo Kα)=9.98 cm-1,F(000)=1080,R1=0.0390,wR2=0.1039;Δρmax=654 e·nm-3,Δρmin=-800 e·nm-3。 配合物的主要键长和键角列于表1,分子结构见图1。
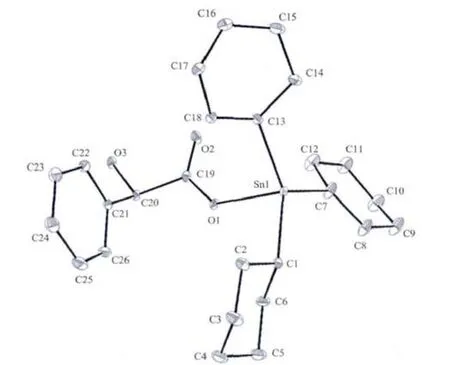
图1 配合物的分子结构图Fig.1 Molecular structure of the title complex

表1 配合物的主要键长和键角Table 1 Selected bond distances(nm)and selected bond angles(°)of the title complex
从图1和结构参数可知,中心锡原子与3个环己碳原子及一个羧基氧原子相连形成四面体构型。由于与R-扁桃酰基的空间效应引起了3个环己基处于不同的空间环境,使得环上C-C键在0.142 5~0.154 4 nm之间形成不对称六元环,且四面体的键角均偏离了正四面体角。3个Sn-C键键长分别为Sn(1)-C(1)0.2160(4)nm、Sn(1)-C(7)0.2143(6)nm 和Sn(1)-C(13)为 0.2113(10)nm,较为接近,与中心锡原子间的键角分别为 C(1)-Sn(1)-C(7)115.6(3)°、C(1)-Sn(1)-C(13)为 121.1(3)°、C(7)-Sn(1)-C(13)120.3(3)°,都比正四面体键角大;Sn-O键键长为0.212 9(3)nm,与Sn(1)-C 键的键角分别为 C(1)-Sn(1)-O(1)为 89.26(18)°、C(7)-Sn(1)-O(1)为 100.6(3)°、C(13)-Sn(1)-O(1)97.4(2)°,都比正四面体角小;这种键角的变化是由于环己基的空间位阻所致。这种空间排列就决定了中心锡与环己基碳原子和氧原子构成畸型四面体。

图2 晶体中的一维链状结构Fig.2 One-dimensional chain structure in the crystal
在晶体中2个三环己基锡R-扁桃酸酯分子间Sn原子和羟基O原子之间的距离为0.264 7 nm,形成了分子间的Sn…O弱键,这样三环己基锡R-扁桃酸酯分子就通过Sn…O弱键的联接而形成如图2所示的一维链状结构。
2.2 配合物的能量和前沿分子轨道组成
根据晶体结构的各原子坐标位置,运用Gaussian03W程序的B3lyp/Lanl2dz基组,在P4计算机上进行量子化学单点计算。配合物计算涉及70原子,349个原子基函,897个初始高斯函数,112个α电子,112个β电子。
结构单元的整体稳定性与体系总能量和前沿轨道的能量密切相关,算得该分子结构单元的总能量ET=-1243.014 678 49 a.u.,EHOMO=-0.228 33 a.u.,ELUMO=-0.030 67 a.u.,可见总能量和占有轨道能量级均较低,最高占有轨道与最低未占有轨道的能量间隙只有0.19766 a.u.,表明结构稳定。从氧化还原或电荷转移的角度分析,最高占有轨道为绝对值较大的负值,|EHOMO|越大说明从HOMO上电离电子越困难,因此,配合物较难失去电子,其基态较稳定。
为探索配合物的成键特征,对配合物分子轨道进行系统分析,用参与组合的各类原子的轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。把配合物原子分为四部分:(a)配体碳原子和氧原子L;(b)环己基碳原子Cy;(c)Sn原子;(d)H原子,前沿占有轨道和未占有轨道各取5个,计算结果见表2和图3。

表2 配合物的分子轨道组成(B3lyp/Lanl2dz)Table 2 Calculated some frontier molecular orbitals composition of the complex at Lanl2dz level

图3 配合物的前沿分子轨道示意图Fig.3 Schematic diagram of frontier MOfor the complex
在最高占有轨道组成中,配体原子对HOMO的贡献总计为97.86%;其他原子对HOMO的贡献则相对较少,锡原子占HOMO成分0.25%;环己基原子1.88%。
在最低未占有轨道组成中,各原子对LUMO的贡献发生较明显变化,锡原子对LUMO的贡献达48.50%;环己基碳原子对 LUMO 的贡献 34.96%;配体碳原子和氧原子贡献为14.87%;氢原子贡献为1.68%。
比较HOMO与LUMO的轨道成份,发现配合物从基态向激发态电子转移时,主要是配体碳原子和氧原子向锡原子和环己基碳原子轨道转移,形成电荷转移配合物。
2.3 配合物的热稳定性分析
利用TG209F3热分析仪,在N2气氛,升温速率为20℃·min-1的实验条件下测定了配合物的热重曲线。结果显示随温度的升高,配合物在274~318℃区间经过热失重,对应于配合物分子失去1个配体扁桃酸根和3个环己基,最后稳定在约28.78%,残余物为SnO2(理论值29.02%)。上述热分析结果表明该配合物结构在274℃之前是可以稳定存在的。
2.4 配合物的环荧光性质
在室温下,用甲醇分别配置浓度约为1×10-5mol·L-1的配体和配合物的溶液,测试了荧光光谱,如图4所示。配体R-扁桃酸在激发波长为445 nm时,最大发射峰出现在536 nm左右,荧光强度较弱,属于配体的π→π*跃迁发光。配合物在激发波长为520 nm时,最大发射峰出现在546 nm左右,和配体的荧光峰相比有一定的红移(10 nm),但荧光强度显著增强。可认为配合物的荧光来自于配体,是一种金属离子微扰的配体发光现象。

图4 配体及配合物的荧光光谱图Fig.4 Fuorescence spectrum of the title complex and ligand
2.5 配合物的电化学性能
采用常规三电极体系:玻碳电极为工作电极,铂电极为辅助电极,SCE为参比电极,以乙醇为溶剂,配合物的浓度为 1.0μmol·L-1, 于室温下进行电化学性质测定。在 -1.000~1.000 V范围内,以 100 mV·s-1速度进行循环伏安扫描,所得结果如图5。从图可看出,配合物无明显氧化还原峰,说明配合物在电极上的电子转移较为困难或者配合物本身较为稳定在常规实验条件下难以被氧化。
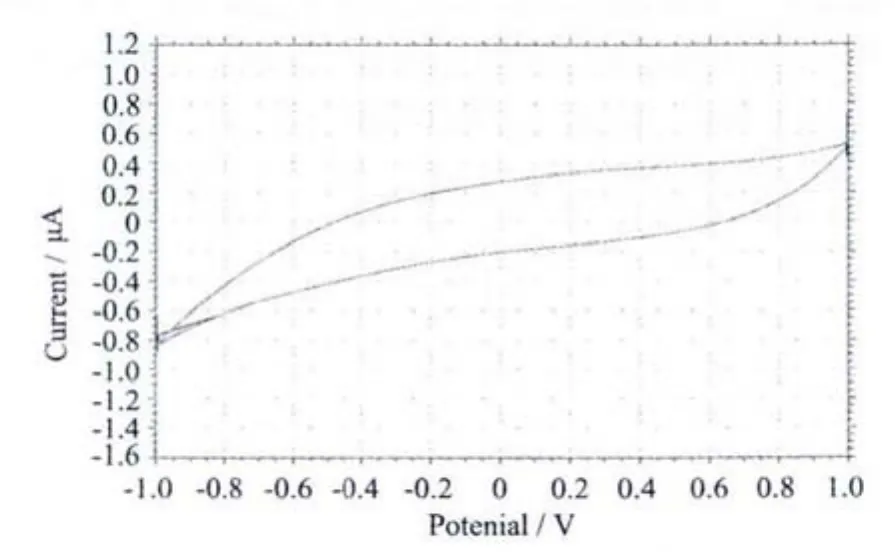
图5 配合物的循环伏安图Fig.5 Cyclic voltammograms of the title complex
[1]Chandrasekhar V,Thirumoorthi R,Metre R K,et al.J.Organomet.Chem.,2011,696:600-606
[2]Effendy,Marchetti F,Marinelli A,et al.J.Inorg.Chem.Acta.,2011,366,388-393
[3]Hanif M,Hussain M,Ali S,et al.J.Polyhedron.,2010,29:613-619
[4]ZHANG Xiao-Yan(张晓燕),YANG Guang(杨 光),ZHANG Jun(张 俊),et al.Chem.J.Chinese Univ ersuties(Gaodeng Xuexiao Huaxue Xuebao),2010,31(6):1162-1166
[5]Siddiqi ZA,Shahid M,Kumar S,et al.J.Organomet.Chem.,2009,694:3768~3774
[6]Ruan B F,Tian Y U,Zhou H P,et al.J.Chim.Acta.,2011,365:302-308
[7]ZHANG Fu-Xing(张复兴),KUANG Dai-Zhi(邝代治),WANG Jian-Qiu(王 剑 秋),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2006,22(7):1321-1326
[8]YAN Wen-Hua(闫文华),KANG Wan-Li(康万利),LI Jin-Huan(李金环).Chinese J.Appl.Chem.(Yingyong Huaxue),2007,24(6):660-664
[9]Shujha S,Shah A,Rehman Z U,et al.Eur.J.Med.Chem.,2010,45:2902-2911
[10]ZHANGFu-Xing(张复兴),KUANGDai-Zhi(邝代治),WANG Jian-Qiu(王 剑 秋 ),et al.Chinese J.Org.Chem.(Youji Huaxue),2008,28(8):1457-1461
[11]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝 代 治 ),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(6):1195-1199
[12]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝 代 治 ),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(9):1890-1894.
[13]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(8):1591-1595
[14]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(6):1111-1115
[15]YU Jiang-Xi(庾江喜),KUANG Dai-Zhi(邝代治),YIN Du-Lin(尹笃林),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(8):1507-1510
[16]ZHANGFu-Xing(张复兴),WANGJian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(2):213-217
[17]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese.J.Appl.Chem.(Yingyong Huaxue),2009,26(6):662-666
[18]Zhang F X,Wang JQ,Kuang D Z,et al.Chinese J.Struct.Chem,2010,29(10):1529-1535
