川芎嗪长循环脂质体包封率的测定
2013-09-14陈道阳陈晓丹王利胜
陈道阳 陈晓丹 王利胜
1.广东省人民政府机关门诊部,广东广州 510030;2.广州中医药大学中药学院,广东广州 510006
川芎嗪是从中药川芎中提取的一种主要有效成分,具有扩张血管、抑制血小板聚集、防止血栓形成、改善微循环等多种药理活性[1],临床主要用于治疗动脉粥样硬化、冠心病、脑血管病、肺动脉高压等疾病[2]。长循环脂质体是一种具有多功能的靶向药物载体,其在体内可阻止巨噬细胞对脂质体的识别和摄取,而极大延长脂质体在血液中的循环时间[3]。把川芎嗪制成长循环脂质体制剂,可显著改善川芎嗪口服吸收差、生物利用度低、半衰期短、代谢快等问题,有效提高了川芎嗪的疗效。本实验旨在建立简单可行、稳定可靠的分析方法,以准确测定川芎嗪在制剂中的含量和包封率,为评价和控制川芎嗪长循环脂质体的质量提供依据。
1 仪器与试药
高效液相色谱仪:Waters515泵、2487紫外检测器(美国Waters公司);N3000色谱工作站(浙江大学);精密分析天平AUW120D(日本岛津公司);JB-3型定时恒温磁力搅拌器(上海雷磁新泾仪器有限公司);微孔滤膜(德国ME MBRANA公司)。
TMP(含量>98.5%,江苏南京泽朗医药科技有限公司);大豆卵磷脂(上海泰伟药业);胆固醇(上海伯奥生物科技有限公司);PEG3000-DSPE(德国lipoid公司);葡聚糖凝胶-G50(上海蓝季科技发展有限公司);磷酸氢二钠、磷酸二氢钠(天津市福晨化学试剂厂);甲醇为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1 样品的制备
2.1.1 川芎嗪长循环脂质体[4]采用乙醇注入法制备,精密称取处方量的川芎嗪、卵磷脂、胆固醇和PEG3000-DSPE,加入适量的无水乙醇使之溶解。将上述乙醇溶液匀速注入到水浴60℃的PBS 6.5溶液中,恒温快速搅拌至无醇味,静置,过0.45 μm的微孔滤膜,即得。
2.1.2 空白脂质体 按处方精密称取卵磷脂、胆固醇、PEG 3000-DSPE等辅料,同法制备不含药物的空白脂质体,备用。
2.2 分析方法的建立[5]
2.2.1 色谱条件 色谱柱:迪马platisil ODS柱(5 μm,250 mm×4.6 mm),Phenomenex保护柱;流动相:甲醇-0.5%冰乙酸(40∶60);流速:1 mL/min;柱温:室温;检测波长:298 nm;进样量:10 μL。
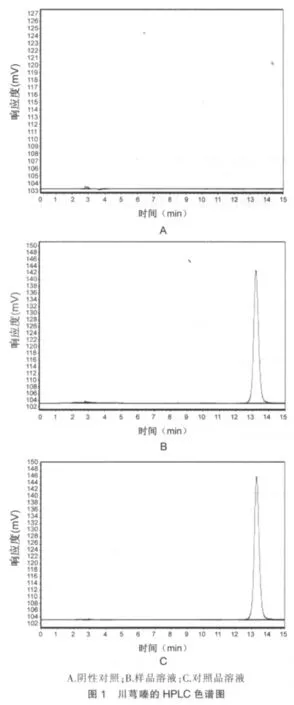
2.2.2 专属性试验 取空白脂质体和川芎嗪长循环脂质体各0.5 mL,置10 mL容量瓶中,加甲醇破乳并定容至刻度,摇匀,分别作为阴性溶液和样品溶液。将阴性溶液、样品溶液和43.60 μg/mL的川芎嗪对照品溶液分别进行HPLC分析。色谱图见图1,川芎嗪的保留时间约为13.2 min,峰形良好,空白辅料对川芎嗪的测定无干扰。
2.2.3 标准曲线的绘制 精密称取川芎嗪对照品8.72 mg,用甲醇配制成浓度为 1.09、2.18、5.45、10.90、21.80、43.60 μg/mL的对照品溶液,按上述色谱条件测定峰面积。以峰面积A为纵坐标,对照品溶液浓度C(μg/mL)为横坐标,绘制标准曲线。得到线性回归方程:A=24 178.21C-101.40(r=0.9995),表明川芎嗪在 1.09~43.60 μg/mL 浓度范围内线性关系良好。
2.2.4 精密度试验 取10.90 μg/mL川芎嗪对照品溶液,连续进样6次,测定峰面积,计算得到RSD为1.41%,说明该法精密度良好。
2.2.5 稳定性试验 取川芎嗪长循环脂质体适量,用甲醇破乳并稀释,摇匀,于室温放置 24 h,分别于 0、2、4、6、8、10、12、24 h进样分析,记录峰面积,计算得到RSD为1.79%,说明样品溶液在24 h内基本上稳定。
2.2.6 重复性试验 取同一批号的川芎嗪长循环脂质体5份,按“2.2.2”项下配制样品溶液,进行分析,结果川芎嗪质量浓度稳定,RSD为1.26%,符合要求。
2.2.7 回收率试验 精密量取 2.18、10.90、43.60 μg/mL的川芎嗪对照品溶液1 mL,分别加入空白脂质体1 mL,加甲醇破乳并定容至10 mL,配制成低、中、高3个浓度的混合溶液各5份,进行液相色谱分析,计算回收率,分别为98.55%(RSD=1.59% ),97.64% (RSD=1.37% ),99.14% (RSD=1.78%),结果表明该分析方法的回收率符合要求。
2.3 分离方法的建立[6]
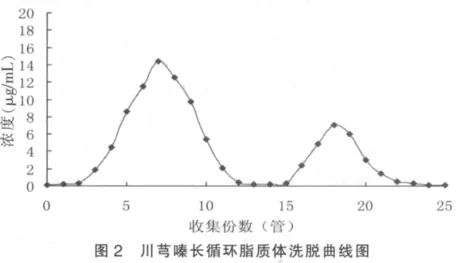
2.3.1 洗脱曲线的绘制 精密量取川芎嗪长循环脂质体0.25 mL,上葡聚糖凝胶-G50微型柱(15 cm×0.8 cm),以PBS 6.8为洗脱液,流速为0.5 mL/min,每毫升为1管收集洗脱液,共收集25管。可见,乳白色的脂质体富集于3~10管洗脱液中。将每管洗脱液用甲醇定容至2 mL,摇匀,过0.22 μm微孔滤膜,测定川芎嗪的含量。以药物质量浓度为纵坐标,收集份数为横坐标,绘制洗脱曲线。图2结果表明川芎嗪长循环脂质体于3~12 mL被洗脱出,而末包封的游离药物于15~23 mL被全部洗脱出来,中间有3 mL间隔,确保了脂质体与游离药物能够完全分离。
2.3.2 柱回收率测定 精密量取2.18、10.90、43.60 μg/mL的川芎嗪对照品溶液0.25 mL上柱,按上述条件进行洗脱,收集15~23 mL的洗脱液,稀释,定容,过0.22 μm微孔滤膜,HPLC法分别测定药物浓度,计算川芎嗪游离药物的上柱洗脱回收率。结果低、中、高浓度的川芎嗪柱回收率分别为97.41% (RSD=1.24% ),99.01% (RSD=1.51% ),96.80%(RSD=1.39%),表明葡聚糖凝胶-G50对川芎嗪没有吸附作用,在以上条件下药物可被完全洗脱。
2.3.3 川芎嗪长循环脂质体包封率的测定[7]精密量取川芎嗪长循环脂质体混悬液0.25 mL,缓缓加于葡聚糖凝胶-G50微型柱(15 cm×0.8 cm)顶部,用 PBS 6.8以 0.5 mL/min的流速进行洗脱,以1 mL/管为单位收集3~12管的洗脱液,合并,加甲醇破乳并定容至18 mL,混合均匀,过0.22 μm微孔滤膜,HPLC测定脂质体中包封的药量。另取川芎嗪长循环脂质体混悬液0.25 mL,用甲醇破乳并稀释定容至5 mL,摇匀,过0.22 μm微孔滤膜,进样分析,测定的川芎嗪的含量为包封和未包封的药物总量。按以下公式计算包封率:包封率=脂质体中包封的药物/药物总量×100%。三批川芎嗪长循环脂质体样品的包封率测定结果见表1。
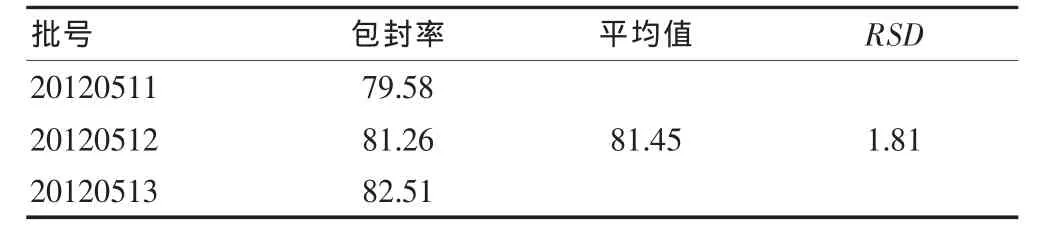
表1 川芎嗪长循环脂质体的包封情况(%)
3 讨论
3.1 测定方法的选择
脂质体包封率的测定方法有很多,在前期试验中曾采用超速离心法,在转速20 000 r/min以上长时间离心,但脂质体仍难以完全沉淀分离,而且在高转速条件下部分脂质体可能被破坏而导致药物渗漏;透析法虽能有效分离,但操作耗时过长[8]。相比之下,凝胶柱色谱法简单可靠,能使包封药物和游离药物得到较好的分离。
3.2 葡聚糖凝胶柱分离条件
本研究选用葡聚糖凝胶G-50能达到较好的分离效果。在分离过程中,色谱柱的径高比、洗脱液的流速及上样量均会影响脂质体与游离药物的分离效果[9]。宜选择能达到最大分离程度的洗脱条件,才能保证包封率测定的准确性。通过前期筛选试验,选择径高比为15 cm×0.8 cm,上样量为0.25 mL,洗脱液为PBS 6.8,流速为0.5 mL/min的洗脱条件,能使川芎嗪长循环脂质体可与游离药物达到最大程度分离。
3.3 流动相的选用
本文建立了准确、稳定的液相色谱分析方法,在选择流动相条件时,若单纯以甲醇-水作流动相,会导致峰形拖尾严重,可能是由于川芎嗪碱性氮原子易与柱上未完全封闭的-OH产生氢键结合[10],使其难以洗脱,造成拖尾。因此参考文献[11-12]在流动相中加入了少量冰乙酸,以调整峰形,达到了良好的效果。
[1]周昌奎,吴晓华.川芎嗪临床应用及研究进展[J].海峡药学,2004,16(6):3-5.
[2]胡国芬,王建平.川芎嗪的药理作用及临床应用进展[J].中国药物与临床,2006,6(10):77.
[3]曹纯洁.长循环脂质体的研究进展[J].药学实践杂志,2005,23(1):1-3.
[4]宏伟,陈大为,赵秀丽,等.姜黄素长循环脂质体的制备及评价[J].中国中药杂志,2008,33(8):889-892.
[5]王利胜,陈晓丹,吕耿斌,等.自乳化辅料对川芎嗪缓释固体分散体释放度的影响[J].中国新药杂志,2012,21(23):96-99.
[6]黄义昆,梁建成,韦敏,等.影响盐酸川芎嗪脂质体包封率各因素分析[J].中国药师,2006,9(9):825-827.
[7]鲁会侠,冯锁民.紫杉醇脂质体药物包封率的测定[J].中国新药杂志,2007,16(10):778-780.
[8]冯秀珍,丁燕飞,李新,等.高效液相色谱法测定槲皮素纳米脂质体药物含量及包封率[J].药物分析杂志,2008,28(4):556-558.
[9]高晓黎,季兴梅.葡聚糖凝胶柱色谱法测定脂质体包封率的条件筛选[J].中国药学杂志,2003,28(7):515-517.
[10]桂双英,温志强,彭代银,等.汉防己甲素脂质体包封率的测定[J].安徽中医学院学报,2009,28(5):70-72.
[11]王利胜,石宗丰,郭琦,等.川芎嗪、阿魏酸微透析体外回收率及体内稳定性的研究[J].中成药,2011,33(2):304-308.
[12]夏祖猛,王利胜,赖宝林,等.芎冰微乳在新西兰兔体内的药动学研究[J].中国实验方剂学杂志,2011,17(11):119-121.
