常染色体显性局灶节段肾小球硬化一家系INF2 基因及临床特征
2013-09-10李国民高学武翟亦晖张晓娥方晓燕利刘海梅
李国民 高学武 徐 虹 翟亦晖 沈 茜 张晓娥 方晓燕 安 宇 孙 利刘海梅 陈 径 曹 琦 饶 佳
局灶节段肾小球硬化( FSGS) 是一种肾小球损伤的常见病理类型,以持续性肾病综合征或非选择性蛋白尿、高血压、镜下血尿和对糖皮质激素耐药为特点,可快速进展为终末期肾病( ESRD)[1~3]。FSGS 分为家族性和散发性,家族性FSGS 又分为常染色体显性( AD-FSGS) 和常染色体隐性( AR-FSGS) 。AR-FSGS 的主要致病基因为NPHS2、NPHS1和PLCE1[4~10]。AD-FSGS 的 主 要 致 病 基 因 为 WT1、ACTN4、TRPC6 和INF2[11~16],极少数由CD2AP 基因突变引起[17,18]。AD-FSGS 和AR-FSGS 致病基因在中国有散发性FSGS 病例的突变报道,但在家族性FSGS 研究中尚未见相关报道。本文以1 个中国AD-FSGS 家系为研究对象,对WT1、ACTN4、TRPC6 和INF2 基因进行直接测序,总结ADFSGS 主要临床特征、基因突变特点,提高对该病的认识。
1 临床资料
1.1 先证者 女,12 岁1 个月,因“体检发现蛋白尿3 d”于2012 年5 月于复旦大学附属儿科医院( 我院) 肾脏科就诊。既往体健,无发热、尿频、尿急和尿痛,无水肿和少尿。肾功能检查正常。肌电图检测正常。其父有类似病史并经病理确诊为FSGS。家族中无高血压和糖尿病病史。鉴于体检发现蛋白尿和其父FSGS 病史,怀疑FSCS 并进一步行家系追踪调查。
1.2 家系调查 本研究征得先证者家系所有成员或成员监护人的书面同意,并经过我院伦理委员会同意(2012032) 。图1 显示1 个上海地区4 代家系图谱,先证者为Ⅳ6。家系成员间无近亲婚配史。该家系共27 名成员,Ⅰ1、Ⅰ2、Ⅱ3、Ⅱ5 死因不详,现有成员23 名,Ⅱ6、Ⅱ8、Ⅲ8、Ⅲ10 和Ⅳ6 为发病者。表1 显示,5 例FSCS 患者中女性3例,男性2 例,平均发病年龄26.9 岁。均以蛋白尿起病。先后3 次对健在的家系成员进行尿常规和尿微量蛋白检测,18 例家族成员尿液分析未见异常;4 例( Ⅱ8、Ⅲ8、Ⅲ10和Ⅳ6) 尿液分析为蛋白尿,1 例( Ⅱ6) 因无尿未能进行尿液分析。Ⅱ8、Ⅲ8 和Ⅲ10 曾在外院进行肾组织活检,Ⅱ8、Ⅲ8 病理检查确诊为FSGS,Ⅲ10 病理表现为局灶硬化伴系膜增殖。Ⅱ6 FSCS 发病10 年已进展为ESRD。Ⅱ8、Ⅲ8 和Ⅲ10 曾在外院行肌电图检测均正常。发病者给予血管紧张素转换酶抑制剂/血管紧张素Ⅱ受体阻滞剂( ACEI/ARB)治疗,能减轻蛋白尿,未给予免疫抑制剂治疗,2 例糖皮质激素治疗无效。
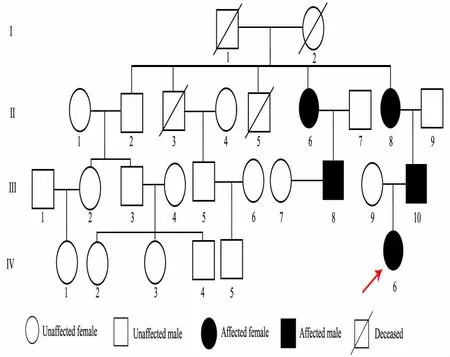
图1 AD-FSGS 家系图谱Fig 1 Pedigree of the AD-FSGS family
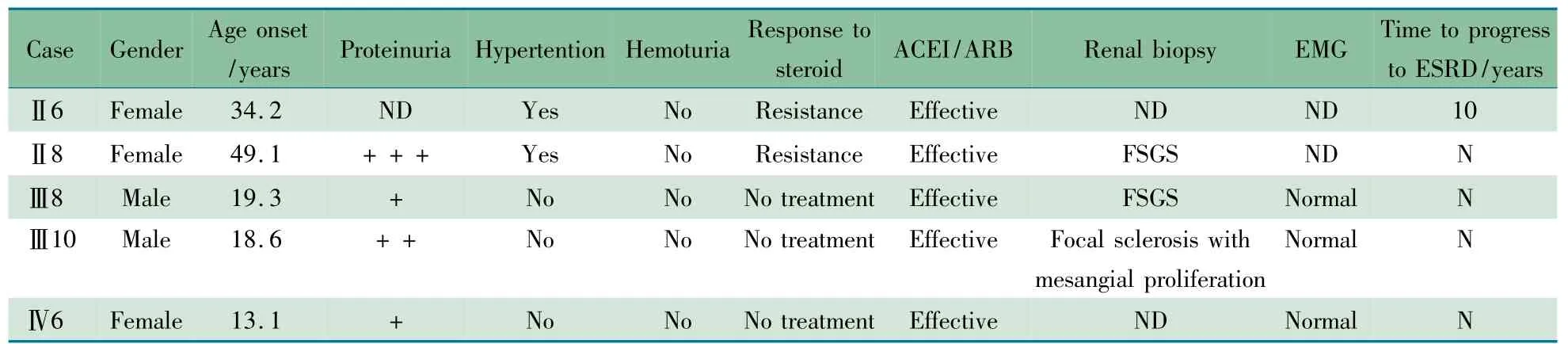
表1 家系中5 例AD-FSGS 患者临床特征Tab 1 Clinical features of patients with AD-FSGS
1.3 诊断 家系中Ⅱ、Ⅲ和Ⅳ各代均有FSCS 发病者,无性别差异,符合常染色体显性遗传特征。先证者家系中有2 例病理诊断为FSGS,1 例为局灶硬化伴系膜增殖,1 例为ESRD,符合家族性/遗传性FSGS 的Duke 诊断标准[19]。3例发病者肌电图检查均正常排除腓骨肌萎缩症。因此,先证者连续3 次尿蛋白+,结合明确的FSCS 家系,诊断为AD-FSGS[20]。
1.4 WT1、ACTN4、TRPC6 和INF2 基因检测 考虑到既往研究对尿液筛查正常者进行基因检测的突变率极低,本研究选取尿常规和尿微量蛋白检测异常的2 个家庭进行基因检测,其中Ⅱ7 和Ⅲ7 拒绝进行基因检测。采用QIAGEN 公司的血液基因组DNA 抽提试剂盒提取7 名家系成员基因组DNA。PCR 扩增WT1 基因外显子8、9,以及ACTN4、TRPC6 和INF2 基因所有外显子,扩增引物序列参考已报道的文献[13~16,18,21]。PCR 反 应条 件: 95℃预 变 性3min,94℃30s,55 ~60℃35 s,72℃40 ~50 s;35 个循环,72℃再延伸5 min。PCR 产物经1%琼脂糖凝胶电泳初步鉴定扩增条带是否为目的条带,若为目的条带则进行测序。使用ABI 3730 测序列分析仪( 美国ABI 公司) 进行测序,使用Chromas 软件读取序列,测序结果与GenBank 中参考序列进行比对,查找突变位点。在7 名家系成员中WT1、ACTN4和TRPC6 基因外显子测序均未发现致病性突变位点,其中5 例发病者( Ⅱ6、Ⅱ8、Ⅲ8、Ⅲ10 和Ⅳ6) INF2 基因外显子8均发现有缺失突变,缺失的碱基为CCCCACCCCCAC,缺失在外显子8 第274 ~285 位点( 图2A) ,未发现INF2 基因其他外显子有致病性突变。另外2 名家系成员( Ⅱ9 和Ⅲ9)在5 例发病者INF2 基因缺失突变区未发现有相似突变( 图2B) ,其他外显子也未发现致病性突变。
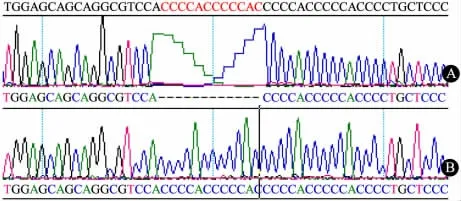
图2 INF2 基因突变图Fig 2 Mutation of INF2 gene
1.5 生物信息学分析 利用美国在线NCBI ( The National Center for Biotechnology Information,http: //www. ncbi. nlm.nih.gov/pubmed) 初步分析突变对编码蛋白结构和功能的影响。INF2 表达分子inverted formin,人源性和鼠源性inverted formin 有高度同源性(80.8%氨基酸相同) 。鼠源性inverted formin 有4 个重要的结构域( 图3) ,即2 个formin 同源结构域( FHl 和FH2) 、1 个Diaphanous 自调节结构域( Diaphanous-autoregulatory domain,DAD) 和1 个Diaphanous 抑制结构域( Diaphanous-inhibitory domain,DID) 。DID 由INF2 基因外显子2 至8 编码,外显子8 上的缺失 CCCCACCCCCAC ( c. 1249delCCCCACCCCCAC,p.T420_P423del) 位于DID( 图3) ,导致DID 结构域构象改变影响inverted formin 功能。
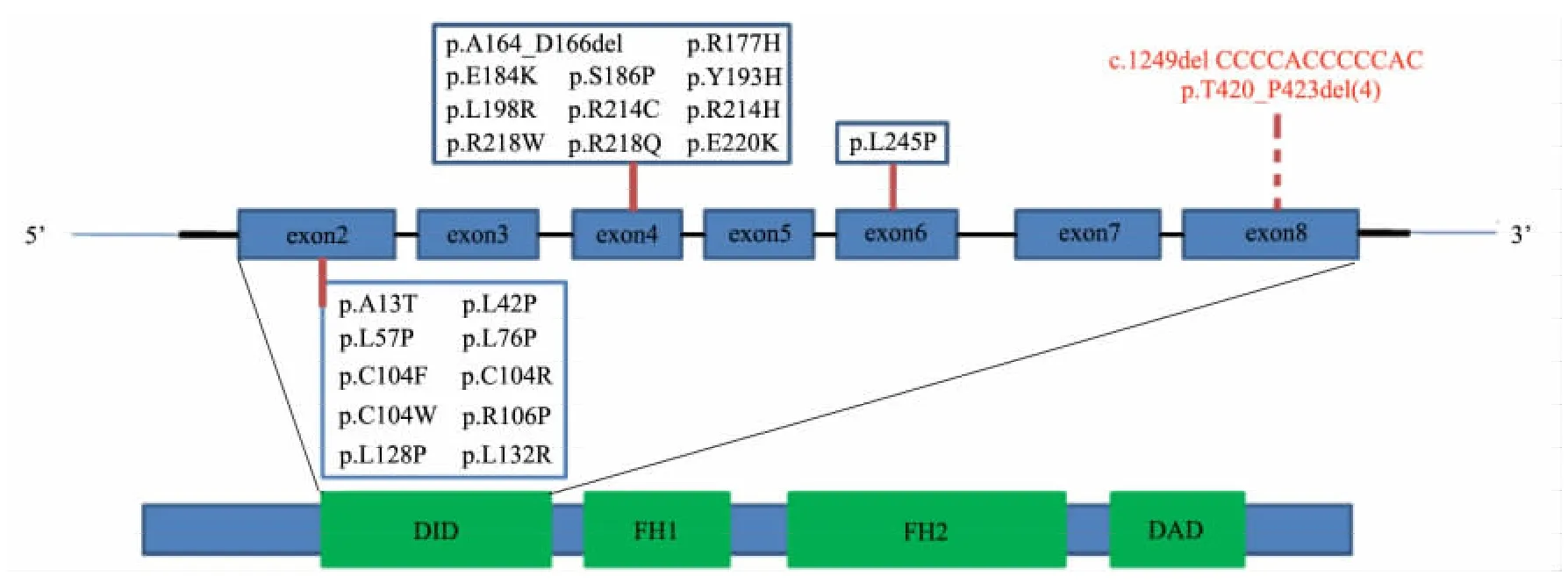
图3 INF2 蛋白结构图及本家系患者缺失突变部位Fig 3 Schematic representation showing exons 2 -8 of the INF2 gene and INF2 protein domain structure
2 讨论
家族性FSGS 多数为遗传性的,由相关致病基因突变引起,占FSGS 总数的18%[1,19]。AR-FSGS 患者临床特征为发病年龄早,一般在婴儿期或儿童早期发病,肾功能进展快速,通常3 ~8 年进展为ESRD,多数表现为肾病综合征[5,10,22,23]。AD-FSGS 患者临床特征为发病年龄晚,一般在青少年或成年期发病,肾功能进展缓慢,可以表现为蛋白尿或肾病综合征,也可伴有血尿和高血压[13,15]。AD-FSGS和AR-FSGS 患者绝大多数对糖皮质激素耐药,对免疫抑制剂不敏感,少数对神经钙调蛋白抑制剂部分有效[5,13,15,22,23]。ACEI/ARB 治疗能减轻蛋白尿和延缓肾功能进展[3]。
本家系图谱分析显示,每代均有患者分布,无性别差异,符合常染色体显性遗传特征。患者平均发病年龄26.9岁,5 例发病者均以尿检异常( 蛋白尿) 就诊,不伴有水肿,糖皮质激素治疗的2 例患者表现耐药,除1 例发病10 年后进展为ESRD,其他暂未进展为ESRD。3 例肾活检患者中,2 例表现为FSGS。本家系患者临床特点与AD-FSGS 患者临床特征相似,符合AD-FSGS 诊断。但本家系是以Ⅳ6 尿液筛查异常为线索,然后追问家族史得以发现,并通过肌电图检测排除腓骨肌萎缩症,最终将Ⅳ6 及其他患者诊断为AD-FSGS。AD-FSGS 发病年龄可以在青少年或成年,存在差异,尽管本家系发病者存在Ⅱ~Ⅳ代发病年龄逐渐年轻的变化( 表1) ,可能与Ⅱ、Ⅲ代发病者出现临床症状较重时才就诊,使发现蛋白尿年龄较迟。本家系的发现警示临床医生对单纯性蛋白尿患者应该追踪其家族其他成员的患病情况。尽管目前通过尿液筛查仅发现家族4 例成员有症状,但不能排除有血缘关系其他成员无病,应该定期随访,定期性尿液筛查,以便早发现、早干预。
本研究通过对WT1、ACTN4、TRPC6 和INF2 基因直接测序发现,该AD-FSGS 家族5 例发病者均存在INF2 基因缺失突变,而且位于外显子8 上。本家系发病者未发现WT1、ACTN4 和TRPC6 基因有致病性突变,可以排除WT1、ACTN4 和TRPC6 基因致病可能。INF2 基因突变可累及外周神经和( 或) 肾脏,引起腓骨肌萎缩症和( 或) FSGS[24]。近年来研究显示,约15%的AD-FSGS 由INF2 基因突变引起[25,26]。早期研究发现INF2 基因突变都位于外显子2 ~4上,主要突变为错义突变,因此对外显子2 ~4 测序可作为INF2 基因突变的筛查方法[27]。但随着研究的深入,在INF2 基因外显子2 ~4 以外也有突变的报道[27]( 图3) 。本研究通过直接测序发现,该AD-FSGS 家族5 例发病者均存在INF2 基因缺失突变,而且位于外显子8 上。故c.1249delCCCCACCCCCAC( p. T420_P423del) 与既往关于INF2 基因研究的报道不同。该突变位于INF2 编码分子inverted formin 重要的结构域DID[27]( 图3) 。该突变可能会引起DID 结构域的改变,影响到inverted formin 对足细胞肌动蛋白调控作用,是足细胞损伤的原因,也是导致本家族AD-FSGS 患者发病的原因。
[1]D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med,2011,365(25):2398-2411
[2]Canetta PA, Radhakrishnan J. Impact of the National Institutes of Health Focal Segmental Glomerulosclerosis (NIH FSGS)clinical trial on the treatment of steroid-resistant FSGS.Nephrol Dial Transplant,2013,28(3):527-534
[3]Gbadegesin R, Lavin P, Foreman J, et al. Pathogenesis and therapy of focal segmental glomerulosclerosis: an update.Pediatr Nephrol,2011,26(7):1001-1015
[4]Philippe A, Nevo F, Esquivel EL, et al. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome.J Am Soc Nephrol,2008,19(10):1871-1878
[5]Kestilä M, Lenkkeri U, Männikkö M, et al. Positionally cloned gene for a novel glomerular protein-nephrin-is mutated in congenital nephrotic syndrome. Mol Cell,1998,1(4):575-582
[6]Frishberg Y, Rinat C, Megged O, et al. Mutations in NPHS2 encoding podocin are a prevalent cause of steroid-resistant nephrotic syndrome among Israeli-Arab children. J Am Soc Nephrol,2002,13(2):400-405
[7]Benoit G, Machuca E, Antignac C. Hereditary nephrotic syndrome: a systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr Nephrol,2010,25(9):1621-1632
[8]Boyer O, Benoit G, Gribouval O, et al. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J Med Genet,2010,47(7):445-452
[9]Jefferson JA, Shankland SJ. Familial nephrotic syndrome:PLCE1 enters the fray. Nephrol Dial Transplant,2007,22(7):1849-1852
[10]Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet,2000,24(4):349-354
[11]Khurana S, Chakraborty S, Lam M, et al. Familial focal segmental glomerulosclerosis (FSGS)-linked alpha-actinin 4(ACTN4) protein mutants lose ability to activate transcription by nuclear hormone receptors. J Biol Chem,2012,287(15):12027-12035
[12]Weins A, Kenlan P, Herbert S, et al. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol,2005,16(12):3694-3701
[13]Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science,2005,308(5729):1801-1804
[14]Brown EJ, Schlöndorff JS, Becker DJ, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet,2010,42(1):72-76
[15]Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4,encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet,2000,24(3):251-256
[16]Ruf RG, Schultheiss M, Lichtenberger A, et al. Prevalence of WT1 mutations in a large cohort of patients with steroidresistant and steroid-sensitive nephrotic syndrome. Kidney Int,2004,66(2):564-570
[17]Benoit G, Machuca E, Nevo F, et al. Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol,2010,25(3):445-451
[18]Gigante M, Pontrelli P, Montemurno E, et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant,2009,24(6):1858-1864
[19]Winn MP. Approach to the evaluation of heritable diseases and update on familial focal segmental glomerulosclerosis. Nephrol Dial Transplant,2003,18(S6):14-20
[20]D'Agati V. Pathologic classification of focal segmental glomerulosclerosis. Semin Nephrol,2003,23(2):117-134
[21]Heeringa SF, Möller CC, Du J, et al. A novel TRPC6 mutation that causes childhood FSGS. PLoS One,2009,4(11):7771
[22]Kitamura A, Tsukaguchi H, Maruyama K, et al. Steroidresistant nephrotic syndrome. Kidney Int,2008,74(9):1209-1215
[23]Caridi G, Bertelli R, Carrea A, et al. Prevalence, genetics,and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulosclerosis.J Am Soc Nephrol,2001,12(12):2742-2746
[24]Boyer O, Nevo F, Plaisier E, et al. INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med,2011,365(25):2377-2388
[25]Boyer O, Benoit G, Gribouval O, et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol,2011,22(2):239-245
[26]Gbadegesin RA, Lavin PJ, Hall G, et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int,2012,81(1):94-99
[27]Sanchez-Ares M, Garcia-Vidal M, Antucho EE, et al. A novel mutation, outside of the candidate region for diagnosis,in the inverted formin 2 gene can cause focal segmental glomerulosclerosis. Kidney Int,2013,83(1):153-159
