婴幼儿杜氏型肌营养不良临床表现及其基因和骨骼肌病理特征
2013-09-10韩春锡林婧娴吴维青陈盼盼谢建生廖建湘
韩春锡 林婧娴 吴维青 陈盼盼 谢建生 廖建湘
杜氏型肌营养不良( DMD) 是以肌纤维变性、坏死、再生及间质结缔组织增生为主要病理特点,以进行性肌无力,肌肉萎缩为主要临床特征的X 连锁隐性遗传性疾病[1,2]。大量肌纤维的坏死,使肌纤维内的CK、AST 和ALT 释放到血液中,导致血清肌酶升高[3,4]。婴幼儿DMD 的运动发育尚未成熟,皮下脂肪丰富,缺乏肌无力、肌萎缩等典型临床表现,大多仅表现为血清CK、AST 或ALT 水平升高,往往被误诊为“心肌炎”或“肝炎”,实施不必要的辅助检查,包括创伤性肝穿刺等[5~9],失去早期诊断、治疗和接受遗传咨询的良机[10,11]。本研究通过分析婴幼儿DMD 的临床表现、血生化、基因以及骨骼肌病理特征,为早期诊断婴幼儿DMD 提供依据。
1 方法
1.1 DMD 的诊断标准 目前尚缺乏婴幼儿DMD 的诊断标准,但鉴于DMD 基因突变检测可为临床诊断提供直接和可靠的依据[2,12],DMD 骨骼肌病理改变是引起临床症状、体征的基础[2,13]。本研究中诊断婴幼儿DMD,至少满足以下2 项中任意1 项方可诊断:①DMD 基因发生移码突变,同时伴有运动发育迟缓、走路不稳和易摔倒等临床表现;②DMD 基因突变不伴有DMD 临床表现时,须通过肌活检证实dystrophin 完全缺失。
1.2 病例纳入标准 深圳市儿童医院( 我院) 从2009 年6月起对在神经肌肉病专科门诊就诊的患儿进行随访观察,故本研究纳入2009 年6 月至2013 年3 月在专科门诊首诊、未在外院接受过糖皮质激素治疗、通过DMD 基因检查和( 或) 肌活检确诊为DMD 的男性婴幼儿。
1.3 分组 为观察患儿运动发育与血清CK、AST、ALT 之间的关系,本研究依据患儿首诊年龄分为4 组:0 ~6、~12、~24 和~36 月龄组。
1.4 血清CK、AST 和ALT 检测 首诊时采集非抗凝静脉血2.0 mL,于我院常规检测CK、ALT 和AST 水平,正常值分别为:CK 25 ~175 U·L-1,AST 0 ~40 U·L-1,ALT 0 ~40 U·L-1。
1.5 肌电图及神经传导速度检查 使用Nicolet 肌电图仪( Viking ⅣP,美国) ,检测正中神经、尺神经、腓总神经的运动、感觉神经传导速度和肱二头肌、股内肌、腓肠肌的同心圆针EMG,包括安静状态下插入电位、自发电位,轻收缩时运动时限、波幅和多相波百分比以及重收缩时运动单位的募集相型和波幅。
1.6 DMD 基因检查[12,14]采集静脉血2.0 mL,EDTA 抗凝,磁珠吸附法提取基因组DNA( Lab-Aid 基因组DNA 分离试剂盒,厦门致善生物科技有限公司) ,MLPA 法( MLPA P034/P035 试剂盒,MRC-HOLLAND 公司) 检测DMD 基因79 个外显子的缺失或重复。对未发现外显子异常的患儿进行DMD 全基因测序,检测点突变。
1.7 肌肉活检[2,15]对非移码缺失突变、重复突变、点突变以及虽是移码缺失突变,但缺乏DMD 临床症状的患儿进行肌肉活检。在利多卡因局部麻醉下取左腓肠肌组织0.5 cm×0.5 cm ×0.6 cm,制成8 μm 厚冰冻切片,实施常规组织化学和组织酶学染色,采用抗dystrophin-C 末端、N-末端、中央棒状区的单克隆抗体1∶100 稀释( Novocastra 公司,美国) ,实施免疫组织化学染色。病理切片由我院神经肌肉病研究室2 名专业医生读片。
1.8 随访 根据患儿年龄,每3 个月至1 年来我院门诊随访,6 月龄起随访运动发育状况。
1.9 统计学方法 采用SPSS 16.0 软件进行统计学分析。计量资料采用x±s表示,组间比较采用方差分析,方差不齐时采用Wiilconxon Signed Ranks 检验。P <0.05 为差异有统计学意义。
2 结果
2.1 一般情况 共纳入男性DMD 患儿43 例,其中门诊患儿17 例,住院患儿26 例。就诊年龄1 ~34( 18.2 ±11.3)月龄。0 ~6 月龄组7 例,~12 月龄组9 例,~24 月龄组8例,~36 月龄组19 例。
对36 例患儿进行了总体运动发育评估,其中31 例(86.1%) 出现运动发育延迟,平均(8.6 ±2.4) 个月时会独坐,(18.6 ±4.9) 个月时会独立行走。对24 例患儿进行了粗大和精细运动发育评估,其中20 例( 83.3%) 在运动发育过程中不会或不愿意爬行运动,行走时左右摇摆,呈鸭步,易跌倒;与同年龄幼儿相比,不能双脚蹦跳,扶着东西上楼梯,地上捡东西时不完全弯曲膝关节,单手或双手扶着膝盖缓慢站立。对19 例患儿进行了语言能力评估,其中5 例(26.3%) 表现为吐字不清。
2.2 就诊原因 43 例DMD 患儿中,因上呼吸道感染,查血清CK 和转氨酶水平升高就诊者分别有21 例( 48.8%)和13 例(30.2%) ,其中误诊为心肌炎14 例,原因不明的高CK 血症7 例;因入托体检时发现ALT 升高误诊为肝炎就诊13 例( 30. 2%) ; 因 单 纯 运 动 发 育 迟 缓 就 诊6 例(14.0%) ;有DMD 家族史并伴有运动障碍,高度怀疑DMD就诊3 例(7.0%) 。
2.3 肌力和肌容积的改变 4 个年龄组的上肢肌力及0 ~6、~12 月龄组患儿的下肢肌力均基本正常。~24 月龄组6/9 例和~36 月龄组所有患儿的下肢近端肌力均为Ⅴ-( 未能蹲起自如) 。~24 和~36 月龄组患儿均有股外侧肌和( 或) 腓肠肌的假性肥大及不同程度的肌肉硬度改变。所有年龄组患儿均未见明显肌萎缩。
2.4 血清CK、AST 和ALT 水平 首诊时43 例DMD 患儿血清CK、AST 和ALT 水平均明显高于正常值。~12、~24和~36 月龄组血清CK 和ALT 水平较0 ~6 月龄组升高( P分别为0.02、002 和0.001) ,~12、~24 和~36 月龄组间血清CK、AST 和ALT 水平差异均无统计学意义( 均P >0.05)( 表1) 。
2.5 DMD 基因检测 43 例DMD 患儿均采用MLPA 法检测DMD 基因79 个外显子的缺失或重复,其中移码缺失突变29 例(67.4%) ,非移码缺失突变2 例( 4.6%) ,单纯重复突变4 例(9.3%) ,重复和缺失同时存在1 例( 2.3%) 。7 例(16.3%) 未发现外显子缺失或重复,实施DMD 全基因测序,均发现有害点突变。
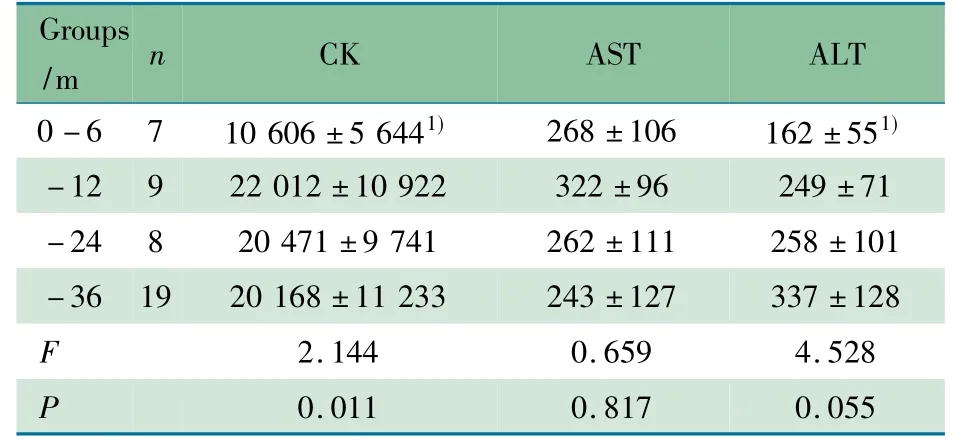
表1 婴幼儿DMD 患儿不同年龄组血清CK、AST、ALT 水平( U·L -1) 比较( x±s)Tab 1 Comparison of serum CK,AST,ALT activity( U·L -1) among different infant DMD groups( x±s)
2.6 肌肉活检 16 例进行肌肉活检。光镜下均可见大量萎缩、变性、坏死肌纤维及再生肌纤维( 图1A) ,间质脂肪结缔组织增生( 图1B) ,符合肌营养不良样病理改变,但很少见到特大的肥大肌纤维、断裂肌纤维以及环状改变的肌纤维。坏死肌纤维周围均可见不同程度的炎细胞浸润。16例患儿肌纤维膜上抗肌萎缩蛋白-C、N 单克隆抗体的免疫染色均表达缺失( 图1C ~E) ,偶见抗肌萎缩蛋白-R 示弱阳性。免疫染色阳性对照抗肌萎缩蛋白表达正常( 图1F) 。
2.7 肌电图及神经传导速度检查 12 例进行了肌电图检查,神经传导速度均正常,其中8 例表现为放松时未见失神经电位,轻收缩时运动单位平均时限变窄、波幅降低,强收缩时呈混合相,提示肌源性损害。
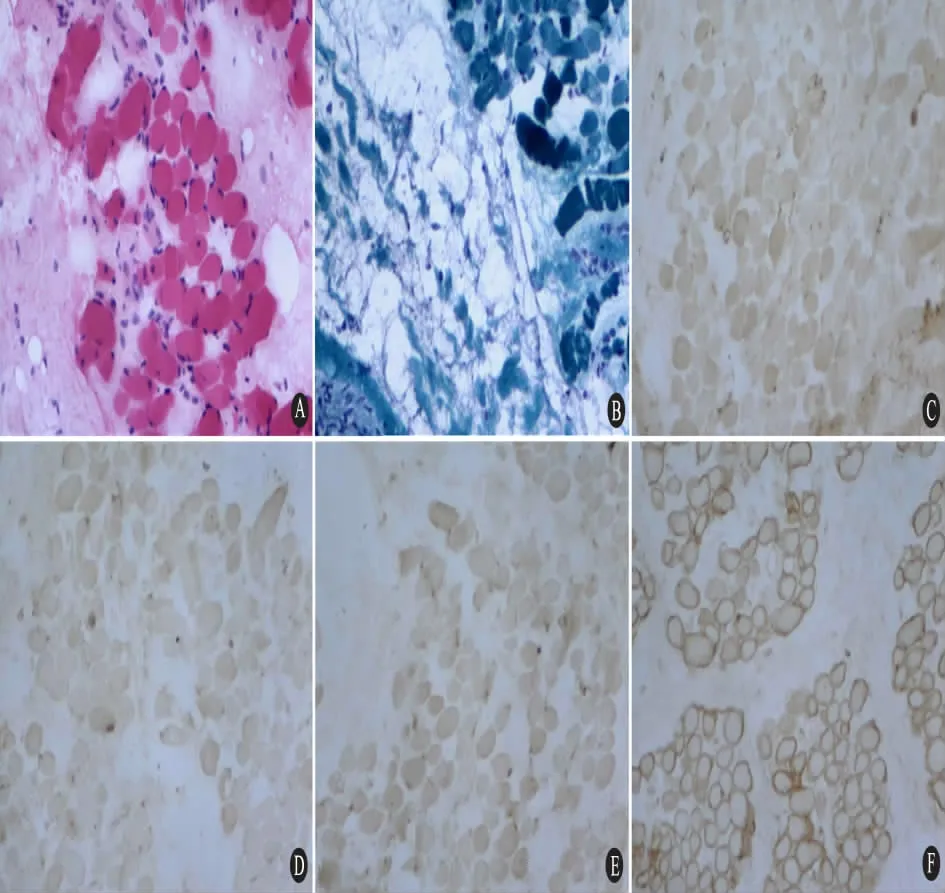
图1 DMD 患儿骨骼肌病理变化和抗肌萎缩蛋白免疫染色( ×200)Fig 1 Pathological changes of skeletal muscle and immunohistochemical staining with antibodies to dystrophin ( ×200)
3 讨论
DMD 患儿的骨骼肌损伤是由躯干和四肢近端开始缓慢发展,表现为进行性、对称性肌无力,肌萎缩伴腓肠肌的假性肥大,下肢受累重于上肢[1,2]。由于髂腰肌和下肢近端肌肉受累早且重,因此患儿由仰卧位站立时,用手支撑躯干呈俯跪位,接着以双手顺次支撑双足背、膝部等处,方能站立,即Gower 征阳性。然而,婴幼儿期DMD 患儿常缺乏典型的阳性Gower 征,仅表现为开始端坐、独站和行走的年龄延迟,行走时两侧摇摆,呈鸭步或行走易跌倒,或从地上捡东西时不全屈曲膝关节,仅用弯腰动作来完成。多数患儿不能或不愿意爬行,而这些临床表现均提示患儿的骨盆带肌和下肢近端肌肉受累,但常未能引起患儿家属或医生的重视,被误认为患儿的行为习惯异常或佝偻病的表现[5,9]。本研究发现,生后1 ~2 个月的DMD 婴儿虽然缺乏肌无力和肌萎缩表现,但在扶着患儿站立状态下可触及股外侧肌和腓肠肌的肌肉硬、韧,是早期诊断DMD 的重要体征,应引起临床医生的重视。如果婴儿早期出现肌无力、肌张力低下和肌萎缩等典型的神经肌肉病表现,则不应考虑DMD,而应考虑先天性肌病、先天性肌营养不良、糖原累积病以及脑和脊髓疾病引起的松软儿等[16]。
肌电图检查有助于鉴别肌源性损伤或神经源性损伤。但在婴儿DMD 的肌电图检查中,因患儿的运动单位电位幅度小,检查不配合,因此要单纯鉴别骨骼肌是否发生神经源性损害还是肌源性损害,肌电图的检查意义不如检测血清CK、AST 和ALT 水平,血清CK、AST 和ALT 成比例升高,直接提示骨骼肌发生肌源性损害,但心肌酶正常不能排除骨骼肌的肌源性损害。一般情况下,骨骼肌发生神经源性损害时,心肌酶正常或轻微升高。
本研究中,患儿就诊原因为CK 和AST、ALT 升高占79%。然而,冯善伟等[17]对436 例( 平均年龄为9.7 岁)DMD 患儿的临床研究结果表明,就诊主要原因为走路困难易摔倒(42%) 和上楼梯困难(38%) ,提示不同年龄段患儿的就诊原因各不相同,不明原因的血清CK、AST、ALT 升高是诊断婴幼儿DMD 的重要线索[18,19]。由于骨骼肌组织中均含有丰富的CK、AST 和ALT,因此当发生肌纤维组织损伤时血清CK、AST 和ALT 均明显升高,其中以CK 的变化最为敏感[20,21]。本研究结果显示,0 ~6 月龄组血清CK 水平超过正常值上限的50 倍以上,而~12 月龄组血清CK 水平可达正常值上限的140 倍。提示: ①DMD 婴儿早期,虽然尚未出现明显的肌无力、肌萎缩等表现,但患儿体内已开始发生肌纤维的大量破坏;②~12 月龄组血清CK 水平升高最显著,可能与患儿活动量增加有关,但可能还有其他因素参与肌纤维的损伤过程[20~24],有必要进一步探讨; ③婴幼儿DMD 血清CK 水平虽然有波动,但始终维持在较高水平,表明患儿的肌纤维破坏有可能持续发生。
本研究患儿血清CK 水平的升高总是伴随着AST 和ALT 水平的升高,这种变化特点不同于骨骼肌外伤或其他组织细胞损伤,如肝炎、心肌炎等引起的血清酶改变。骨骼肌外伤或心肌炎引起的CK 升高,往往随着病因的消除,其CK 水平也逐渐恢复至正常,同时伴有相应疾病的症状和体征,如骨骼肌外伤史或心脏功能异常以及心电图的改变等。虽然,肝炎患儿也可出现AST 和ALT 升高,但一般不会伴有CK 的明显升高。因此,当发现男性婴幼儿出现原因不明的血清CK、AST 和ALT 伴随升高时,应注意肌营养不良的可能,即使是缺乏DMD 临床症状,也有必要进行DMD基因检测协助诊断[19]。由于DMD 外显子的缺失或重复只占DMD 基因突变的70%左右,因此高度怀疑DMD,但仍未发现外显子的缺失或重复时,应该实施DMD 全基因测序或肌活检。
当发生DMD 基因突变,但并不改变翻译阅读框架( 即非移码突变)[16],则临床上并不表现为典型的DMD,而表现为症状较轻的贝克型肌营养不良( BMD) 。因此,一般情况下只要发生移码突变,不管是外显子的缺失、重复还是点突变,婴幼儿DMD 的临床表现都较类似,表现为运动发育延迟,走路不稳容易摔倒。但部分外显子的非移码突变可导致DMD,详细机制尚不清楚。肌活检是诊断和鉴别诊断肌病的重要手段,但肌活检不是诊断DMD 的绝对适应证[18]。如婴幼儿期已经出现DMD 的临床表现,同时检测到DMD 外显子缺失或重复,并通过DMD 结构与功能相关网站数据库( eDystrophin online database) 查找基因突变属于移码突变,就可确诊DMD[13]。本研究结果表明,DMD 婴幼儿骨骼肌中已经发生肌纤维的大量萎缩、变性、坏死、再生以及脂肪结缔组织增生等较典型肌营养不良的病理改变,提示早期干预婴幼儿DMD 的必要性。
[1]Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1:diagnosis, and pharmacological and psychosocial management.Lancet Neurol,2010,9(1):77-93
[2]杉田秀夫,小泽瑛二郎,梦中征载. 新肌肉病学. 南江堂,东京: 1995.469-486
[3]McMillan HJ, Gregas M, Darras BT, et al. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics,2011,127(1):132-136
[4]Urganci N, Arapoqlu M, Serdaroqlu P, et al. Incidental raised transaminases: a clue to muscle disease. Ann Trop Paediatr,2006,26(4):345-348
[5]Zamora S, Adams C, Butzner JD, et al. Elevated aminotransferases activity as an indication of muscular dystrophy: case reports and review of the literature. Can J Gastroenterol,1996,10(6): 389-393
[6]Veropalumbo C, Del Giudice E, Esposito G, et al.Aminotransferases and muscular diseases: a disregarded lesson.Case reports and review of the literature. Paediatr Child Health,2012,48(10):886-890
[7]Wright MA, Yang ML, Parsons JA, et al. Consider muscle disease in children with elevated transaminase. J Am Board Fam Med,2012,25(4):536-540
[8]Liu P(刘平), Wu J, Hu WG, et al. Analysis of clinical characteristics of 5 children with progressive muscular dystrophy misdiagnosed as having virus hepatitis. Clinical Misdiagnosis &Mistherapy(临床误诊误治),2012,25(7):40-42
[9]Zhao CT(赵春婷), Zhang QZ, Wang T. 肌营养不良误诊为心肌炎一例. New Medicine(新医学),2012,43(6): 424-425
[10]Moat SJ, Bradley DM, Salmon R, et al. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK) . Eur J Hum Genet,2013. Epub ahead of print
[11]Iwańczak F, Stawarski A, Potyrala M,et al. Early symptoms of Duchenne muscular dystrophy--description of cases of an 18-month-old and an 8-year-old .patient. Med Sci Monit,2000,6(3):592-595
[12]Nicolas A, Lucchetti-Miganeh C, Yaou RB, et al. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J Rare Dis,2012,7:45
[13]王德新. 肌肉疾病. 神经病学. 北京:人民军医出版社,2007.214-228
[14]Witting N, Duno M, Vissing J. Becker muscular dystrophy with widespread muscle hypertrophy and a non-sense mutation of exon 2. Neuromuscul Disord,2013,23(1):25-28
[15]Taylor LE, Kaminoh, YJ, Rodesch CK, et al. Flanigan.Quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens. Neuropathology and Applied Neurobiology 2012,38:591-601
[16]Singh SM, Mallela KM. The N-terminal actin-binding tandem calponin-homology (CH) domain of dystrophin is in a closed conformation in solution and when bound to F-actin . Biophys J,2012,103(9):1970-1978
[17]Feng SW(冯善伟), Liang YY, Cao JQ, et al. Duchenne 型假肥大肌营养不良患儿的主要生活事件. J Appl Clin Pediatr(实用儿科临床杂志),2012,27(24):1866-1868
[18]Han CX(韩春锡). Diagnostic approach and muscle biopsy of myopathies in children. J Appl Clin Pediatr(实用儿科临床杂志),2012,27(24):1860-1863
[19]Chang XZ(常杏芝), Yuan Y, Qin J. Persistent hypertransaminasemia as the presenting finding of childhood occult muscle disease. Chinese Journal of Medicine(中国医刊),2006,41(4):42-44
[20]Kohli R, Harris DC, Whitington PF. Relative elevations of serum alanine and aspartate aminotransferases in muscular dystrophy. J Pediatr Gastroenterol Nutr,2005,41(1):121-124
[21]Zamora S, Adams C, Butzner JD, et al. Elevated aminotransferases activity as an indication of muscular dystrophy: case reports and review of the literature. Can J Gastroenterol,1996,10(6): 389-393
[22]Lev EI, Tur-Kaspa I, Ashkenazy I, et al. Distribution of serum creatine kinase activity in young healthy persons. Clin Chem Acta,1999,279(1/2):107-115
[23]Meltzer H. Factors affecting serum creatine phosphokinase levels in the general populations: the role of race, activity and sex. Clin Chim Acta,1971,33(1):165-172
[24]Baird MF, Graham SM, Baker JS, et al. Creatine-kinase-and exercise-related muscle damage implications for muscle performance and recovery. J Nutr Metab,2012,2012:960363
