CUDA加速的DNA-蛋白质匹配及其优化
2013-09-07陈春雷慕德俊张慧翔
陈春雷,慕德俊,张慧翔,胡 伟
(西北工业大学 自动化学院,陕西 西安 710072)
结构生物信息学已成为当前计算机科学领域的一个研究热点。其主要研究目标是获取生物系统的高分辨率结构信息,从而精确地推理系统的功能、预测某些修改或扰动对系统造成的影响。与生物信息学的其他领域相比,结构生物信息学面临众多新的挑战。其中之一是多数结构生物信息学问题的搜索空间是连续的;换言之,生物系统的微观结构一般通过原子在空间坐标系中的位置来表示,而坐标通常是连续变量,因此搜索空间是无穷的。虽然某些近似方法可以在一定程度上应对结构生物信息学问题的这种连续特性,但是从搜索空间中求得最终解仍然需要进行大量的运算[1]。
DNA-蛋白质匹配是生物信息学的一个重要问题。传统的序列分析方法往往会导致较高的多重检验错误率 (false-positive rate)[2]。多数基于结构生物信息学的DNA-蛋白质匹配方法把获得转录因子-DNA组合体的信息作为一个必备条件,而转录因子-DNA组合体的信息通常需要通过实验获得,这种实验通常需要耗费大量的时间。参考文献[2]提出的基于退火算法的匹配算法避开了这个问题,但是使用该方法进行DNA-蛋白质匹配仍需要较大的运算量。参考文献[3]基于参考集索引技术提出了一种快速的序列分析算法,并将其应用于DNA-蛋白质匹配。参考文献[4]采用多CPU的方式设计实现了并行化的DNA-蛋白质序列分析方法,并取得了较高的加速比。但上述工作均未涉及基于结构生物信息学的匹配方法的加速问题。
应对大运算量问题的一个常用方法是并行计算。NVIDIA公司统一计算设备架构CUDA(Compute Unified Device Architecture)是一种全新的并行计算架构。在CUDA的支持下,通用计算图形处理单元GPGPU(General Purpose Graphic Processing Unit)可以作为一种通用的效率、硬件加速器,发挥出强大的运算能力[5]。
本文针对参考文献[2]的DNA-蛋白质匹配方法,从计算的角度对其进行分析,设计实现并优化了基于CUDA的加速方法,通过实验验证了其加速性能。
1 相关知识介绍
1.1 基于退火算法的DNA-蛋白质匹配算法
给定一条DNA链和一条蛋白质链,两者之间的相对位置存在很多种可能。假定两者均为刚体,DNA-蛋白质组合体的物理能量主要受两者相对位置的影响,组合体的物理能量越低,其稳定性越强。该方法试图寻找使得组合体最稳定的相对位置,这一结果对生物信息学的研究具有重要意义。
从计算的角度看,该方法的基础是退火算法。为了提高搜索精度,该匹配方法通常需要随机产生大量初始“种子”,为每个“种子”启动一个基于退火算法的搜索过程,在整个解空间中寻找最稳定的相对位置。这种匹配方法的流程如图1所示。

图1 基于退火算法的DNA-蛋白质匹配方法流程图
图1中的初始值为T0,温度T及温度衰减周期interval为正整数,温度衰减系数为a。每当算法所经历的平移和旋转次数达到interval的非零整数倍时,T衰减为原来的a倍(0<a<1),Tmin为温度的最小值。
DNA-蛋白质组合体的物理能量E1、E2分别代表第n次(1≤n≤Smax,Smax为平移和旋转次数的最大值)平移和旋转前后组合体的物理能量。Emin代表通过算法得到的组合体物理能量的最小值。如果E2<E1,则接受第n次平移和旋转的结果;否则,计算指数式exp(-(E2-E1)/T)的值,并使用C++库函数drand48()产生一个在区间[0,1]上均匀分布的随机数。如果指数式的值小于随机数的值,则接受第n次平移旋转的结果,反之不接受。
step为当前进行到了第几次平移和旋转。
1.2 CUDA编程模型及线程调度方式的特点
CUDA程序通常包含CPU串行代码和GPU并行代码,后者被称作CUDA程序的内核。在执行内核时,CUDA会产生大量并行工作的线程,以单指令多数据SIMD(Single Instruction Multiple Data)方式完成整个内核的计算任务。CUDA采用网格(grid)和线程块(block)来组织线程[6]。一个block的线程又被划分为一个或多个线程组(warp)。warp是CUDA程序最基本的调度单位,属于同一个warp的各个线程会从CUDA程序代码段中相同的程序地址同时开始执行,但是各线程具有独立的当前指令指针和寄存器状态。因此,各线程可能会有不同的执行路径,例如执行不同的分支选择结构代码或循环结构代码[7]。warp执行特点如图2所示。

图2 warp的执行特点示意图
假设一个warp中共有4个线程:线程1~线程4,它们的执行时间分别是t1~t4,并且t3<t1<t2<t4。因为CUDA的基本调度单位是一个完整的warp而非单个线程,所以整个warp的执行时间取决于执行时间最长的线程t4。其他3个线程在执行完毕后必须等待还没有执行完毕的线程,因此有一段时间处于空闲状态,相应的计算资源也就被闲置了。例如,线程3闲置的计算资源最多,其空闲时长为(t4-t3)。造成计算资源闲置的原因是同一warp中各个线程的执行路径不同;而CUDA采用的是SIMD并行方式,执行路径的不同是由每个线程所处理的数据差异造成的。因此,依照一定规则对数据进行重新排列是减少这种计算资源闲置状况的常用方法[8]。重排规则通常视具体应用而定。
2 使用CUDA加速DNA-蛋白质匹配
2.1 从计算角度分析匹配算法
匹配方法的流程已由图1给出,除参数初始化外,该方法可分为三部分:(1)平移、旋转DNA和蛋白质;(2)计算DNA-蛋白质组合体的能量;(3)后续处理。这3部分在普通CPU平台上所耗时间依次为∶73.624 s、1138.945 s、110.809 s(仅使用一个随机初始种子,退火算法参数的预设值及软硬件平台参数指标见本文第3部分),实验使用的DNA、蛋白质结构数据来自参考文献[9]编号为2GLI的文件。其他的DNA、蛋白质结构数据的实验结果也显示了计算能量所耗费的时间远超过其他两个部分。计算能量时,DNA-蛋白质组合体所处的三维空间被划分成若干个棱长为埃米(10-10m)级的晶格。一条DNA链由若干个DNA原子构成,一条蛋白质链由若干个蛋白质原子构成;以2GLI组合体为例,共有855个DNA原子和1 270个蛋白质原子。每次平移和旋转前后,每个原子所属的晶格都可能发生改变,每个晶格所包含的原子个数也可能发生改变。Di(i=1,2,…,ND),Pj(j=1,2,…,NP)分别代表组合体中的DNA原子数量和蛋白质原子数量。DNA原子i(i=1,2,…,ND)在三维空间中所处的晶格和相邻的晶格共有27个,统称为原子i的临近晶格(Neighboring Lattice),以pi,js(x,1,2,…,ni)代表这27个晶格中的所有蛋白质原子,这些原子即原子i的相邻蛋白质原子。以di,jx代表DNA原子i与蛋白质原子jx之间的欧式距离,两个原子之间的能量是di,jx的函数:E(di,jx)。则组合体的总能量为:

根据以上分析可知:(1)计算能量是匹配方法中最耗时部分;(2)E(di,jx)(i=1,2,…,ND;x=1,2,…,ni)的计算是相互独立的,无依赖关系,适于并行。根据阿姆达尔定律[10],选择计算能量部分进行并行化。
2.2 基于CUDA的并行匹配算法
以下伪代码给出了串行方式计算能量的步骤:
算法1:串行方式计算能量
(1)E←0
(2)for i=1 to ND
(3) for all neighboring lattice of atomi∶la=1 to 27
(4) for all protein atoms in lattice la
(5) compute E(di,jx)and E←E+E(di,jx)
(6) end for
(7) end for
(8)end for
令CUDA block为一维结构,一个block中包含B个线程,一个初始种子的搜索过程仅由一个block完成。基于以上串行代码,给出CUDA内核的伪代码:
算法2:并行方式计算能量
(1)tid←threadIdx.x
(2)Ethis_thread←0,stride←「ND/B⏋
(3)for s=0 to stride
(4)i←s.B+tid
(5)if i<ND
(6) for all neighboring lattice of atom i∶la=1 to 27
(7) for all protein atoms in lattice la
(8) compute and E(di,jx)and Ethis_thread←Ethis_thread+E(di,jx)
(9) end for
(10) end for
(11)end for
算法2实现了算法1第2行循环操作的并行化。每个CUDA线程对应一个DNA原子,依次进入该原子的27个相邻晶格,找到各个相邻蛋白质原子,计算E(di,jx),并累加到能量的部分和:Ethis_thread。在CUDA内核返回后,通过累加各线程的能量部分和可以得到组合体的总能量。另外,如果B<ND,则需要把ND个DNA原子平均分成「ND/B⏋块(最后一块可能不足B个原子),然后,由整个block的线程依次处理各块,算法2第3行的循环结构即为了达到这个目的。
2.3 优化并行算法
考虑到本文1.2节所述warp的执行特点,上述并行方式存在一定的不足。算法2中第7行的循环次数取决于当前晶格中蛋白质原子的个数,同一warp中各线程的循环次数可能会不同,如果差异过大,会造成严重的计算资源闲置;对所有线程而言,算法2第3行、第6行循环结构的循环次数是确定的。
假设DNA存储在一个数组中,且该数组位于GPU主存(global memory)中。为了尽量减少计算资源闲置,重排DNA原子的原有次序(DNA链中的次序),处于同一个晶格中的DNA原子被存储在数组的某一段连续区域内。采用这种优化措施是基于以下原因:(1)由式(1)可知,总能量由E(di,jx)做算术加法得到,与E(di,jx)的计算次序无关;(2)为了提高GPU主存的带宽利用率,同一warp中的线程通常从主存中的临近区域读取数据,即内存访问对齐 (coalescing)[6];(3)DNA的双螺旋结构是非线性的,DNA在链中相邻的原子未必处于同一晶格中;(4)同一晶格中的DNA原子的临近蛋白质原子数量相同,重排可以减少因线程执行路径差异造成的计算资源闲置。重排的效果如图3所示。

图3 DNA原子存储次序重排示意图
3 实验
3.1 实验平台
硬 件 平 台:Intel®CoreTM2 E7500 CPU,2.93 GHz,NVIDIA GTX 660图形处理器(单个warp包含32个线程),CPU主存4 GB。
软件平台:Linux操作系统内核版本2.6.18-194.el5,gcc版本4.1.2,CUDA版本5.0。
3.2 实验参数设置与结果
DNA-蛋白质结构数据编号为2GLI。CPU版程序以单线程串行方式执行;GPU版程序block size固定为512;各block从不同的初始种子出发,分别基于退火算法进行搜索,每个block对应一个初始种子。退火算法参数:初始温度T0=120℃,最低温度Tmin=0.001℃,温度衰减周期interval=800,温度衰减系数a=0.99,最大平移旋转次数Smax=106。
实验结果如表1所示。与单线程CPU程序相比,未经优化的GPU程序将获得最高可达8倍左右的加速比;而经过重排优化后,加速比在此基础上进一步显著提升,最高可达15倍左右。
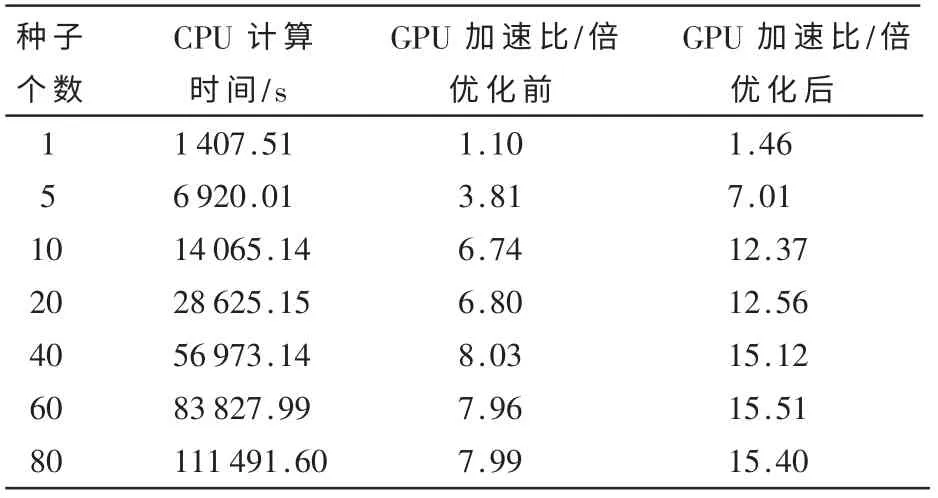
表1 实验结果
本文针对一种典型的DNA-蛋白质匹配算法,设计实现了基于CUDA的并行化方法,从线程调度的角度对该方法进行优化,并通过实验验证了加速性能。实际应用往往需要为一个组合体产生大量的初始种子(数百个),并为每个种子开启一个基于退火算法的搜索过程;其目的是达到较高的匹配精度。实验显示,使用单个GPGPU加速,在单个block包含的线程数不变的前提下,随着初始种子数量的增加,加速比逐渐趋于稳定。例如,当初始种子个数超过40后,加速比基本稳定在15倍左右。其原因在于单个GPGPU的计算能力存在上限,当种子足够多时,其计算能力已得到较充分利用,无法继续提高加速比。为了满足实际应用的需求,下一步的工作将考虑使用基于GPGPU的集群来加速匹配算法,以进一步提高加速比。
[1]BOURNE P E,WEISSIG H.Structural bioinformatics[M].Hoboken∶Wiley-Liss Inc,2003.
[2]Liu Zhijie,Guo Juntao,Li Ting,et al.Structure-based prediction of transcription factor binding sites using a protein-DNA docking approach[J].Proteins,2008,72(4)∶1114-1124.
[3]戴东波,熊赟,朱扬勇.基于参考集索引的高效序列相似性查找算法[J].软件学报,2011(4)∶718-731.
[4]Zhang Zhang,Xiao Jingfa,Wu Jiayan,et al.ParaAT∶A parallel tool for constructing multiple protein-coding DNA alignments[J].Biochemical Biophysical Research Communications,2012,419(4)∶779-781.
[5]徐新海,杨学军,林宇斐,等.一种面向CPU-GPU异构系统的容错方法[J].软件学报,2011,22(10):2538-2552.
[6]NVIDIA Cooperation.CUDA programming guide version 5.0[EB/OL].[2013-05-15].http://docs.nvidia.com/cuda/cudac-programming-guide/.
[7]TIANYI D H,TAREK S A.Reducing branch divergence in GPU programs[A].In Proceedings of the Fourth Workshop on General Purpose Processing on Graphics Processing Units[C].London∶ACM.2012.
[8]IMEN C,AHCENE B,NOUREDINE M.Reducing thread divergence in GPU-based b&b applied to the flow-shop problem[A].In Proceedings of the 9th International Conference on Parallel[C].Berlin∶Springer-Verlag.2011.
[9]Rutgers and UCSD.Protein Data Bank[DB/OL].[2009-02-24].http://www.rcsb.org/pdb/explore/explore.do?structureId=2gli.
[10]GENE A.Validity of the single processor approach to achieving large-scale computing capabilities[A].In Proceedings of the April 18-20(AFIPS′67)[C].New York∶ACM.1967.
