合成1-(1,3-噻唑啉-2-基)-3-巯基氮杂环丁烷盐酸盐的工艺改进*
2013-09-01朱小华陈国华
王 杰,朱小华,陈国华
(1.中国药科大学 药物化学系,江苏 南京 210009;2.南京天海医药科技有限公司,江苏 南京 210009)
替比培南酯{(1R,5S,6S)-6-[(1R)-羟乙基]-1-甲基-2-[1-(1,3-噻唑啉-2-基)氮杂环丁烷-3-基硫基]碳青霉-2-烯-3-羧酸新戊酰氧基甲酯}是日本明治制药株式会社研究开发的世界上第一个口服碳青霉烯类抗生素,是替比培南的酯类前药,口服后在酯酶作用下水解为替比培南,再与细菌青霉素结合蛋白结合,抑制细菌细胞壁的合成。该药于2009年8月在日本首次上市,临床用于儿科肺炎、中耳炎、鼻窦炎,对肺炎球菌、耐大环内酯类药物的肺炎链球菌和耐青霉素的肺炎链球菌有效,商品名为 Orapenem[1,2]。
1-(1,3-噻唑啉-2-基)-3-巯基氮杂环丁烷盐酸盐(1)是合成替比培南酯的关键中间体,目前合成路线主要有3条[3~5]:(1)以二苄胺为原料,经取代、环合、氢化脱苄合成3-羟基氮杂环丁烷盐酸盐,再与2-甲硫基-2-噻唑啉缩合,羟基活化、亲核取代、碱性水解共7步反应制得1,总收率31.4%;(2)以苄胺为原料,经取代、环合、羟基活化、氢化脱苄、亲核取代、氧化、缩合、水解、成盐等8步反应制得1,总收率21%;(3)以烯丙胺为原料,先溴代、环合制得1-氮双环丁烷,再经过乙酰化、水解、缩合共5步反应制得1,总收率44%。方法(2)步骤长,且多步反应使用的原料较昂贵。方法(3)虽然产率高、步骤短,但所用原料毒性较大,且多步反应后处理需要柱层析分离,不利于工业化生产。
本文在文献[4~8]方法的基础上,设计了一条制备1的新路线。以苄胺为原料,与环氧氯丙烷开环生成N-苄基-3-氯-2-羟基丙胺(2);2在碱性条件下经分子内环合得N-苄基-3-羟基氮杂环丁烷(3);3成盐酸盐后加压氢解脱苄基得到4;4经Boc酸酐、甲磺酰氯“一锅法”制得5;5再经硫代乙酸钾亲核取代、酸性水解制得3-巯基氮杂环丁烷盐酸盐(7);7与2-甲硫基-2-噻唑啉缩合合成了1(Scheme 1),其结构经1H NMR,IR和MS确证。其中由5制备7的方法尚未见文献报道。
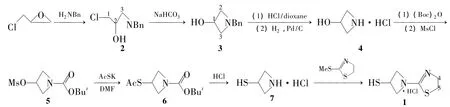
Scheme 1
改进路线具有反应条件较温、操作简便和总收率高(40%)等优点,适合工业化生产。
1 实验部分
1.1 仪器与试剂
RY-1型熔点仪(温度未校正);Bruker AV-300型核磁共振仪(CDCl3为溶剂,TMS为内标);Nicolet Impact 410型红外光谱仪(KBr压片);Agilent 1100 ESI-MS型质谱仪。
所用试剂均为化学纯。
1.2 合成
(1)2的合成
在反应瓶中加入苄胺22.0 g(250 mmol)和水250 mL,搅拌下于0℃加入环氧氯丙烷22.0 g(237.5 mmol),反应 16 h。抽滤,滤饼干燥得粗品,用丙酮/石油醚重结晶得白色晶体2 43.5 g,收率92%,m.p.72℃~73℃(72℃~73℃[4]);1H NMR δ:7.23~7.37(m,5H,ArH),3.88(m,1H,2-H),3.81(s,2H,CH2Ph),3.56(dd,J=9.9 Hz,4.2 Hz,2H,1-H),2.84(dd,J=12.3 Hz,3.6 Hz,1H,3-H),2.72(dd,J=12.3 Hz,7.8 Hz,1H,3-H);MS m/z:200{[M+H]+}。
(2)3的合成
在反应瓶中加入2 44.8 g(225 mmol),碳酸氢钠20.2 g(240 mmol)和乙腈250 mL,搅拌使其溶解,回流反应6 h。冷却至室温,过滤,滤液减压蒸除溶剂,加入石油醚30 mL,于5℃反应1 h。过滤,滤饼干燥得白色固体3 33.7 g,收率92%,m.p.63 ℃~64 ℃(64 ℃~65 ℃[4]);1H NMR δ:7.23~7.33(m,5H,ArH),4.40(m,1H,1-H),3.61(m,4H,2,3-H),2.95(m,2H,CH2Ph);MS m/z:164{[M+H]+}。
(3)4的合成
在反应瓶中加入3 32.6 g(200 mmol)和二氧六环200 mL,搅拌使其溶解;于5℃以下通入干燥氯化氢气体至pH 1后,反应1 h。抽滤,滤饼干燥得白色晶体38 g。将其加入10%Pd/C 5 g和95%乙醇400 mL中,通入H2,于1 MPa/室温条件下反应20 h。过滤,滤液减压浓缩至干,加入甲苯60 mL,于0℃反应1 h。抽滤,滤饼真空干燥得白色晶体4 19.6 g,收率 90%,m.p.88 ℃~90 ℃(86 ℃~88 ℃[6]);1H NMR(DMSO-d6)δ:9.00(brs,2H,NHHCl),6.15(d,J=6.3 Hz,1H,OH),4.50(m,1H,1-H),4.02(m,2H,2-H),3.71(m,2H,3-H);MS m/z:74{[M+1]+}。
(4)5的合成
在反应瓶中加入4 11.0 g(100 mmol),三乙胺20.0 g(200 mmol)和二氯甲烷100 mL,搅拌使其溶解;于0℃以下加入(Boc)2O 24.0 g(110 mmol),反应 3 h。加入三乙胺 10.0 g(100 mmol),于5℃以下滴加甲磺酰氯11.4 g(100 mmol)的二氯甲烷(40 mL)溶液,滴毕,反应2 h。过滤,滤液用饱和碳酸氢钠溶液(2×75 mL)萃取,合并有机层,减压蒸除溶剂得黄色油状物5 21.4 g,收率 85%;1H NMR δ:5.20(m,1H,1-H),4.26(m,2H,2-H),4.08(m,2H,3-H),3.07(s,3H,CH3in Ms),1.44(s,9H,CH3in Bu);MS m/z:252{[M+H]+}。
(5)7的合成
在反应瓶中加入5 15.1 g(60 mmol),AcSK 11.3 g(90 mmol)的DMF 150 mL,搅拌使其溶解;回流反应12 h。减压蒸除溶剂,加入乙酸乙酯200 mL,依次用饱和碳酸氢钠溶液400 mL,饱和食盐水400 mL洗涤,无水硫酸钠干燥,减压浓缩得黄色油状物6 12.5 g(直接用于下步反应);MS m/z:232{[M+H]+}。
在反应瓶中加入6 12.5 g和浓盐酸100 mL,搅拌下于50℃反应10 h;冷却至室温,用乙酸乙酯(2×50 mL)萃取,水层减压浓缩得无色油状物7 5.0 g,收率 92.6%(5→7);1H NMR δ:4.26(m,2H,2-H),4.06(m,3H,1,3-H);MS m/z:90{[M+H]+}。
(6)1的合成
在反应瓶中加入7 5.0 g(55 mmol),2-甲硫基-2-噻唑啉 7.2 g(60 mmol)和碳酸氢钠 8.4 g(110 mmol)的甲醇(80 mL)溶液,搅拌下回流反应6 h。减压蒸除溶剂,残余物用混合溶剂[V(乙腈)∶V(THF)=1 ∶6]重结晶得白色晶体1 7.8 g,收率68%,m.p.132℃~134℃(134℃~136℃[3]);1H NMR δ:12.49(s,1H,HCl),5.26(m,1H,1-H),4.61(t,J=12.7 Hz,2H,4-H),4.11(m,2H,2-H),3.98(m,2H,3-H),3.56(t,J=12.7 Hz,2H,5-H),2.30(d,J=8.5 Hz,1H,SH);IR ν:2 955,2 411,1 635,1 445,1 238,1 135,755,667 cm-1;MS m/z:175{[M-Cl+H]+}。
2 结果与讨论
在中间体3的合成中,方法(1)以二苄胺为原料,价格较贵,且环合后需要柱层析分离,收率仅为48%;本文以价格相对便宜的苄胺代替,第一步中调整投料比例为n(苄胺)∶n(环氧氯丙烷)=1.05 ∶1.00,避免了生成苄胺二取代杂质,环合后无需柱层析分离,3的总收率提高至85%,适合工业生产。
由4制备5中,氨基保护和羟基活化采用“一锅法”,中间体不需分离,反应收率高,操作简便;由5制备7的方法尚未有文献报道;制备6中Ac-SK的亲核取代反应可通过增加AcSK用量并提高温度,使收率提高。由6制备7选择盐酸进行反应,在水解酯键同时脱Boc保护,还使7以盐的形式存在,利于下一步反应处理。
本路线在最后一步才引入N-噻唑啉基团,减少了2-甲硫基-2-噻唑啉总用量,降低了成本与环境危害性。
[1] 黄金竹,母连军.碳青霉烯类抗生素的研究概况[J].国外医药(抗生素分册),2007,28(4):145-154.
[2] 张文君,吴文芳,冯小龙.新型口服碳青霉烯类抗菌药物-泰吡培南酯[J].河北医药,2010,32(18):2596-2598.
[3] Isoda T,Ushirogochi H,Satoh K,et al.Syntheses and pharmacokinetic studies of prodrug esters for the development of oral carbapenem,L-084[J].J Antibiot,2006,59(4):241-247.
[4] Isoda T,Yamamura I,Tamai S,et al.A practical and facile synthesis of azetidine derivatives for oral carbapenem,L-084[J].Chem Pharm Bull,2006,54(10):1408-1411.
[5] Hayashi K,Hiki S,Kumagai T,et al.Synthesis of azetidine derivatives using 1-azabicyclo[1.1.0]butane[J].Heterocycles,2002,56(1):433-442.
[6] Krishna R,Udaykiran D,Chintamani U,et al.Development of an optimized process for the preparation of 1-benzylazetidin-3-ol:An industrially important intermediate for substituted azetidine[J].Org Pro Res Dev,2011,15:462-466.
[7] Cui J,Michelle T D,Shen H,et al.Structure based drug design of crizotinib(PF-02341066),a potent and selective dual inhibitor of mesenchymal epithelial transition factor(c-MET)kinase and anaplastic lymphoma kinase[J].J Med Chem,2011,54:6342-6363.
[8] Stahl G L,Walter R,Smith C W.General procedure for the synthesis of mono-N-acylated 1,6-diaminohexanes[J].J Org Chem,1978,43(11):2285-2286.
