P掺杂锐钛矿相TiO2的第一性原理计算*
2013-08-31郑树凯吴国浩刘磊
郑树凯 吴国浩 刘磊
(河北大学电子信息工程学院,河北大学计算材料研究中心,保定 071002)
1 引言
TiO2具有三种晶体结构:板钛矿、金红石和锐钛矿相.其中,在环境净化领域常用锐钛矿相TiO2光催化降解各种有毒有害物质,这是因为锐钛矿相TiO2的禁带宽度约为3.2 eV,比金红石相TiO2的禁带宽度略大0.2 eV,导致锐钛矿相TiO2中的光生电子和空穴具有更强的还原和氧化能力,从而具有更强的光催化活性.由于板钛矿相TiO2的光催化活性很低,因此较少应用于光催化领域.但是在实际应用当中,较大的禁带宽度也限制了锐钛矿相TiO2光催化能力的发挥,因为只有波长小于约387.5 nm的紫外光才能激发电子从价带跃迁至导带产生具有氧化还原能力的电子-空穴对,导致锐钛矿相TiO2不能充分利用太阳光中的大部分可见光.因此,如何有效地扩展TiO2的光响应范围至可见光区,不仅在实验上而且在理论上已成为光催化研究领域的热点问题[1−3].2001年Asahi等[4]的研究论文指出,N掺杂可有效扩展TiO2的光响应范围至可见光区并使掺杂样品表现出更强的光催化活性,从而掀起了非金属离子掺杂改善TiO2光催化活性的研究热潮.例如,Hamadanian等[5,6]研究发现,相比未掺杂前,S掺杂TiO2的光催化活性有一定程度的提高.Wang等[7]的研究表明,Cl掺杂导致TiO2的吸收边向长波长方向移动,在可见光照射下该样品对邻苯二甲酸丁基苄基酯(BBP)的降解率可达92%.Parayil等[8]发现C修饰的TiO2在模拟的太阳光照射下可以很好地分解水制备氢,表现出比纯TiO2更高的光催化活性.
近年来,在实验上制备非金属P掺杂TiO2的研究较多[9−13],研究结果均表明适量P掺杂可以有效地提高TiO2的光催化活性.如Zheng等[14]的研究表明,P掺杂TiO2在大于400 nm可见光照射下表现出明显的光催化活性.Yu[15]的研究结果显示P掺杂TiO2的光催化降解亚甲基蓝的效率要比商用P25高.但对于P掺杂提高TiO2光催化活性的机理却存在不同解释.如Zheng等[14]认为,与未掺杂TiO2相比,P掺杂TiO2具有更大的比表面积和更小的晶粒尺寸是导致P掺杂提高样品光催化活性的原因,而Yu[15]则认为,掺杂的P元素溶解在TiO2中形成间隙固溶体是光催化提高的重要因素.因此,为了从本质上揭示P掺杂TiO2光催化活性提高的原因,本文运用基于密度泛函理论的第一性原理计算了P不同掺杂形式对TiO2相关性质的影响,并对结果进行了讨论.
2 计算模型
考虑到实验上P掺杂TiO2的晶体结构多为锐钛矿相[9,11,13−15],并且P的最佳掺杂含量为P:Ti=2 at%—3 at%[14,15],因此本文构建了含108个原子的3×3×1的锐钛矿相TiO2超晶胞进行计算.构造P掺杂锐钛矿相TiO2模型时,分别考虑了P原子在锐钛矿相TiO2晶格中存在的三种掺杂形式,即:P替位Ti,P替位O以及P以间隙式原子存在,如图1所示.其中,①位为P替位Ti,②位为P替位O,③位为间隙P.分别用P@Ti-TiO2,P@O-TiO2和P@in-TiO2表示P替位Ti,P替位O以及间隙P存在时的锐钛矿相TiO2.掺杂的P原子均处在所选晶胞的内部,可以尽量减小边界效应的影响.

图1 不同P掺杂形式锐钛矿相TiO2的结构模型
计算采用基于密度泛函理论的平面波超软赝势方法进行,电子与离子实之间的相互作用使用超软赝势进行描述.在相应模型结构优化(含对全部原子位置的弛豫)的基础上计算体系的性质,电子间的交换关联能由广义梯度(GGA)下的PBE(Perdew-Burke-Ernzerhof)泛函描述,平面波的截止能量选择为300 eV,能量收敛标准设为每原子2×10−5eV,第一布里渊区按2×2×2进行分格,参与计算的价电子为:O 2s22p4,Ti 3s23p63d24s2,P 3s23p3.所有计算均在倒易空间中进行.
本文研究人员均为河北大学刘保亭教授课题组成员,全部计算采用该课题组购买的CASTEP软件完成.
3 计算结果与讨论
3.1 结构优化
表1为不同形式P掺杂锐钛矿相TiO2的相应模型结构优化结果.从表1可以知道,实验上获得的纯TiO2的晶格参数[16]与本文中TiO2结构优化后的参数相比很接近,说明本文所选择的模型可靠,计算结果可信.

表1 不同形式P掺杂前后锐钛矿相TiO2的结构参数
不同形式的P掺杂均造成了TiO2晶格的畸变.由于P5+的半径为0.035 nm,比Ti4+的半径0.068 nm小很多[15],因此P替位Ti时TiO2晶胞体积比未掺杂前的小,P替位O和P以间隙原子形式存在时,TiO2的晶胞体积变大.由于P以间隙原子存在时体系多了一个额外的原子,所以此时TiO2晶格膨胀程度最大.Sato等[17]曾经报道,畸变八面体的电偶极矩产生的局域内部场会促进初始光激发产生的载流子的分离,并因此加强体系的光催化活性.由于间隙P的引入使锐钛矿相TiO2的晶格畸变程度最大,由此可以推测间隙P的存在或许是提高TiO2光催化活性的一个重要因素.同时由于P离子半径与Ti离子半径相差较大,实验上制备的阳离子P掺杂TiO2样品,P是很容易以间隙掺杂形式存在的.这也是本文推测间隙P存在是提高TiO2光催化活性的一个重要原因.
3.2 电荷布居
为了考察P所处的化学环境,对不同掺杂形式下的P进行了电荷布居计算,结果如表2所示.从表2可以知道,当P替换Ti时,P的氧化程度最高,P以间隙形式存在时,其也是处于被氧化的状态,价电子部分地转移到O.P替位O时,其只有轻度的还原,这是因为P的电负性比O低,吸引电子的能力较弱所致.

表2 不同掺杂形式P的电荷布居
Peng等[18]利用密度泛函理论的第一性原理计算了P掺杂锐钛矿相TiO2的相关性质.结果表明,在富钛条件下,锐钛矿相中P替位O和P替位Ti的形成能分别为10.52和11.26 eV;在富氧条件下,P替位O和P替位Ti的形成能分别为15.48和1.32 eV;而P在特定的间隙位置,最小的形成能为4.13 eV.从这些数据可以看出,P在富氧条件下是很容易发生P替位Ti的.这也是实验上制备P掺杂TiO2时P多以阳离子P5+形式存在的重要原因.虽然间隙P较之于P替位Ti难于形成,但是X射线光电子能谱(XPS)分析表明[19],P掺杂锐钛矿相TiO2中除了存在P5+之外,还存在P的低价氧化态.而本文的电荷布居结果表明,P以间隙态存在时,其氧化态确实低于P替位Ti时的氧化态.因此,结合Peng等[18]的计算数据,在特定的制备条件下,实验上是很有可能产生间隙P的.
3.3 能带结构
图2(a)—(d)分别是计算得到的不同形式P掺杂前后锐钛矿相TiO2费米能级附近的能带结构,图中始终将电子能够填充的最高能级作为能量零点.计算得到的锐钛矿相TiO2的禁带宽度为2.198 eV,比实验值3.230 eV低,这是由于GGA方法本身所固有的缺点造成的,但该方法计算结果的相对值是非常准确的,并不影响对问题的定性讨论.

图2 计算得到的能带结构 (a)TiO2;(b)P@Ti-TiO2;(c)P@O-TiO2;(d)P@in-TiO2
从图2可以看出,三种情况下P掺杂均导致TiO2的禁带宽度增大,禁带宽度增大的程度为P@in-TiO2(2.389 eV)>P@Ti-TiO2(2.244 eV)>P@O-TiO2(2.242 eV).禁带宽度的增大可以使TiO2的吸收边发生蓝移.这将导致P掺杂锐钛矿相TiO2需要更高能量的光子激发才能使价带电子跃迁到导带产生电子空穴对,而这是对扩展TiO2光响应范围不利的.但是可以注意到,三种P掺杂情况下均在TiO2禁带之内引入了孤立的能级.P@Ti-TiO2的孤立能级位于价带顶上方约0.216 eV(对G点而言)处,该能级距离导带底较远,是电子的深能级俘获陷阱,电子从价带跃迁到该掺杂能级需要的光子能量很小,并且跃迁电子很容易从该掺杂能级返回价带.同时由于费米能级处于导带之内,该能级电子到导带的跃迁不是主要的,主要的光激发跃迁仍然是TiO2的价带到导带,因此P@Ti-TiO2的光学吸收边仍然表现为蓝移.P@O-TiO2的掺杂能级有三条(包括费米能级),均处在TiO2的禁带之内,并且距离价带顶较远.虽然P@O-TiO2的禁带宽度较TiO2大,但由于在TiO2禁带之内的掺杂能级较多,并且体系电子最高填充的能级(即费米能级)在禁带之内,电子受合适能量光子激发时从这些掺杂能级跃迁到导带,导带电子会优先跃迁回这些掺杂能级,因此P@O-TiO2的光学吸收边会表现为红移.同时由于P@O-TiO2的掺杂能级数目较多,很容易形成光生电子的复合中心,降低TiO2的光催化效率.P@in-TiO2的一条掺杂能级位于TiO2禁带之内,约在价带顶上方0.785 eV(对G点来说),该掺杂能级距离价带顶和导带底均较远,可以成为光生电子-空穴对的分离中心,延长光生载流子的寿命,因此可以预期,P以间隙原子形式存在时,P@in-TiO2的光催化活性能够得到很大提高.
由于P替位Ti或以间隙原子存在时表现为阳离子,而实验上测得的P掺杂锐钛矿相TiO2中的P会以P5+和/或P3+存在[14,20],结合本文计算得到的P的电荷布居数,本文认为P替位Ti时,P以P5+形式存在,而P以间隙原子形式存在时,P以P3+的形式存在.因此可以总结得出,实验上制备P掺杂TiO2时,虽然大部分P会以替位Ti形式存在,但是仍然会有间隙形式的P存在.此外,由于P替位O需要很高的形成能[21],实验上获得P@O-TiO2的可能性是比较低的.P掺杂TiO2的光催化活性提高的原因在于替位Ti的P和间隙内的P.实验上获得的阳离子P掺杂TiO2的吸收边发生蓝移[14,22],也与本文的能带计算结果相符合.
3.4 分态密度
图3(a),(b)是锐钛矿相TiO2内部两个O原子和一个Ti原子的电子分态密度图.从图3中可以明显看到TiO2的价带主要由O 2p和Ti 3d轨道构成,而导带则主要由Ti 3d轨道贡献.
图4(a)—(i)为不同P掺杂形式TiO2中各元素原子的分态密度图.为了方便对比,并且由于掺杂原子对邻近的原子态密度影响较大,因此本文取邻近P原子的两个O原子和一个Ti原子做分态密度图.

图3 锐钛矿相TiO2的分态密度 (a)O;(b)Ti
从图4中可以看出,由于不同形式P掺杂的影响,掺杂后TiO2内部原子轨道的杂化有相应的变化:
1)未掺杂前TiO2的O原子主要有三个态密度峰,表明O 2p与Ti 3d轨道杂化后的电子轨道处于简并态;当P替位Ti时,P@Ti-TiO2中的O 2p的态密度峰变成六个,说明由于P的掺入对邻近P的O原子的电子轨道产生了微扰,使原先处于简并态的O 2p轨道各能级分裂;P 3p轨道主要在价带底下方与O 2p和Ti 3d轨道发生了杂化,产生了较强的相互作用;
2)P替位O时,对邻近的Ti原子的电子态密度影响较大,P@O-TiO2中的掺杂能级两条主要由P 3p,Ti 3d和O 2p轨道杂化而成,另外一条主要由Ti 4s和Ti 3p杂化构成,P替位O对体系的影响体现在价带顶上方的掺杂杂化能级上;
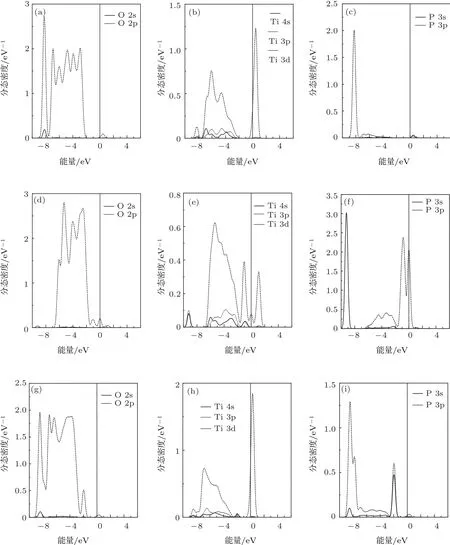
图4 分态密度 P@Ti-TiO2中的(a)O,(b)Ti,(c)P;P@O-TiO2中的(d)O,(e)Ti,(f)P;P@in-TiO2中的(g)O,(h)Ti,(i)P
3)P以间隙原子形式存在时,从态密度峰的位置上看,P对Ti和O的电子态密度均有较大影响,在P@in-TiO2的禁带之内掺杂能级除了由P 3p,Ti 3d和O 2p轨道贡献外,P 3s轨道也参与了杂化,这种情况在P@Ti-TiO2和P@O-TiO2中并未出现,P 3p和O 2p轨道还在P@in-TiO2价带下方发生了较强的杂化现象.
3.5 吸收光谱
图5是计算得到的P掺杂前后TiO2的光吸收曲线,计算时采用了TiO2的多晶模型.由于电子在能带之间和能带内的跃迁频率都远大于声子频率,因此在CASTEP的计算中仅考虑了电子的跃迁,介电函数可以表述为线性响应函数.光学响应函数通常可以由复介电函数 ε(ω)=ε1(ω)+iε2(ω) 来表述,而介电函数的实部和虚部之间的关系可以通过Kramers-Kronig变换关系相联系.根据费米黄金定律,可以从直接跃迁概率的定义推导出介电函数的虚部ε2(ω),结果为

式中u是入射电场的极化方向矢量,V和C分别表示价带和导带,K为倒格矢,为动量跃迁矩阵,和分别为价带和导带上的本征能级.通过介电函数虚部可以得到体系的吸收曲线[23].
从图5中可以看出,相对于未掺杂前,P@Ti-TiO2和P@in-TiO2的光学吸收边均有一定程度的蓝移,P@in-TiO2的蓝移程度比P@Ti-TiO2大.而P@O-TiO2的光学吸收边相比于TiO2则有一定程度的红移.光学吸收边的偏移与本文前面能带结构的计算结果符合较好.一般来说,吸收带边的蓝移减小了TiO2的光响应范围,而红移则扩展了TiO2的光响应范围.因此,光学吸收边红移是TiO2可见光光催化应用的关键.但是,实验上获得的P掺杂TiO2中的P均以阳离子形式存在[9,13,14,20,24,25],并且研究结果均表明合适含量的P掺杂确实提高了TiO2的光催化活性,所以实验上掺杂P提高TiO2光催化活性的原因不是阴离子P造成的.由此推测,P掺杂提高TiO2光催化活性的原因应该是P替位Ti或间隙P造成的.结合图5中的吸收光谱结果可以认为,在紫外光下P掺杂提高TiO2光催化活性[10,11,14,15,25]的原因在于吸收边的蓝移导致光生电子空穴对具有更强的氧化还原能力,而可见光下P掺杂提高TiO2光催化活性[9,13]的原因则主要来源于间隙P的存在使体系对可见光具有更强的吸收能力.因此,间隙P的存在应该是TiO2光催化活性提高的一个重要原因.本文的计算结果支持了Yu[15]对P掺杂提高TiO2光催化活性的研究,即考虑到P和Ti离子的半径相差较大,P应该是进入TiO2中形成了间隙固溶体的推测.

图5 P掺杂前后TiO2的吸收曲线
4 结论
采用基于密度泛函理论的第一性原理方法计算了P替位Ti,P替位O以及P以间隙原子存在时锐钛矿相TiO2的晶格参数、P的电荷布居、能带结构、态密度和吸收光谱.计算结果表明,P替位Ti时晶胞体积变小,P替位O和间隙P掺杂使TiO2晶胞体积变大.替位Ti的P和间隙P均为正的化合价,而替位O的P由于电负性较小,仅有轻度的还原.不同P掺杂形式均使锐钛矿相TiO2的禁带宽度变大,但由于掺杂形式不同导致P@Ti-TiO2和P@Ti-TiO2的吸收边蓝移,P@O-TiO2的吸收边红移.计算结果支持了P可能以间隙形式存在于TiO2晶格中形成固溶体从而提高TiO2光催化活性的实验结果.
[1]Zhang Y,Tang C Q,Dai J 2005 Acta Phys.Sin.54 323(in Chinese)[张勇,唐超群,戴君2005物理学报54 323]
[2]Peng L P,Xia Z C,Yang C Q 2012 Acta Phys.Sin.61 127104(in Chinese)[彭丽萍,夏正才,杨昌权2012物理学报61 127104]
[3]Wu G H,Zheng S K,Liu L,Jia C J 2012 Acta Phys.Sin.61 223101(in Chinese)[吴国浩,郑树凯,刘磊,贾长江2012物理学报61 223101]
[4]Asahi R,Morikawa T,Ohwaki T,Aoki K,Taga Y 2001 Science 293 269
[5]Hamadanian M,Reisi-Vanani A,Behpour M,Esmaeily A S 2011 Desalination 281 319
[6]Hamadanian M,Reisi-Vanani A,Majedi A 2010 Appl.Surf.Sci.256 1837
[7]Wang X K,Wang C,Jiang W J,Guo W L,Wang J G 2012 Chem.Eng.J.189–190 288
[8]Parayil S K,Kibombo H S,Wu C M,Peng R,Baltrusaitis J,Koodali R T 2012 Inter.J.Hydrogen Energy 37 8257
[9]Lin L,Lin W,Xie J L,Zhu Y X,Zhao B Y,Xie Y C 2007 Appl.Catal.B:Environ.75 52
[10]Lv Y Y,Yu L S,Zhang X L,Yao J Y,Zou R Y,Dai Z 2011 Appl.Surf.Sci.257 5715
[11]Jin C,Qiu S C,Zhu Y X,Xie Y C 2011 Chin.J.Catal.32 1173
[12]Asapu R,Palla V M,Wang B,Guo Z H,Sadu R,Chen D H 2011 J.Photochem.Photobio.A:Chem.225 81
[13]Shi Q,Yang D,Jiang Z Y,Li J 2006 J.Mol.Catal.B:Enzym.43 44
[14]Zheng R Y,Lin L,Xie J L,Zhu Y X,Xie Y C 2008 J.Phys.Chem.C 112 15502
[15]Yu H F 2007 J.Phys.Chem.Solids 68 600
[16]Xu L,Tang C Q,Qian J,Huang Z B 2010 Appl.Surf.Sci.256 2668
[17]Sato J,Kobayashi H,Inoue Y 2003 J.Phys.Chem.B 107 7970
[18]Peng Y H,He J F,Liu Q H,Sun Z H,Yan W S,Pan Z Y,Wu Y F,Liang S Z,Chen W R,Wei S Q 2011 J.Phys.Chem.C 115 8184
[19]Lin L,Lin W,Zhu Y X,Zhao B Y,Xie Y C 2005 Chem.Lett.34 284
[20]NakahiraA,KonishiK,YokotaK,HonmaT,AritaniH,TanakaK2006 J.Ceram.Soc.Jpn.114 46
[21]Baunack S,Oswald S,Scharnweber D 1998 Surf.Interface Anal.26 471
[22]Körsi L,Dékány I 2006 Colloid Surf.A:Physicochem.Eng.Asp.280 146
[23]Xiong S Y,Wang L,Dong Q M,Liang P 2012 Sci.China:Phys.Mech.Astron.42 6(in Chinese)[熊斯雨,王乐,董前民,梁培2012中国科学:物理学力学天文学42 6]
[24]Zhang Y Y,Fu W Y,Yang H B,Liu S K,Sun P,Yuan M X,Ma D,Zhao W Y,Sui Y M,Li M H,Li Y X 2009 Thin Solid Films 518 99
[25]Lv Y Y,Yu L S,Huang H Y,Liu H L,Feng Y Y 2009 J.Alloys Compd.488 314
