RP-HPLC法同时测定苯甲酸原料药中有关物质苯甲醛和甲苯的含量
2013-08-29章家伟王辉孙庆荣王向峰雷玉萍帅放文湖南尔康制药股份有限公司长沙410331湖南省药用辅料工程技术研究中心长沙410331湖南省食品药品检验研究院长沙410001
章家伟,王辉,孙庆荣,王向峰,雷玉萍,帅放文#(1.湖南尔康制药股份有限公司,长沙410331;2.湖南省药用辅料工程技术研究中心,长沙 410331;3.湖南省食品药品检验研究院,长沙 410001)
苯甲酸是羧基直接与苯环碳原子连接形成的最简单的芳香酸,又称安息香酸,其在医药、食品、化工等方面应用十分广泛,可用于合成制备多种药物、防腐剂以及生产苯酚等。
苯甲酸收录于2010年版《中国药典》(二部)[1]、《欧洲药典》(EP)7.0版、《美国药典》(USP)34-《国家处方集》(NF)29版、《英国药典》(BP)2010年版以及《日本药局方》(JP)16版中,但各国药典均未对苯甲酸有关物质进行检查,也尚未见文献报道苯甲酸有关物质的测定方法。
苯甲酸的工业生产方法主要为甲苯液相空气氧化法[2],此外,还有三氯甲苯水解法以及邻苯二甲酸酐脱羧法等[3]。以甲苯为原料合成苯甲酸,其杂质主要为副产物苯甲醛以及残留反应物甲苯等。目前,文献报道的苯甲醛的检测方法主要有气相色谱法[4-5]、电位滴定法[6]和分光光度法[7]等;甲苯的检测方法主要为气相色谱法[4]。近年来,已有文献报道采用高效液相色谱(HPLC)法检测苯甲醛[8]以及甲苯[9]。在前人工作基础上,笔者参考文献资料[10-11],建立了反相(RP)HPLC法同时测定苯甲酸有关物质的方法。结果表明,所建立的方法简便、快捷、结果准确,能有效控制药品质量。
1 材料
1.1 仪器
LC-15C HPLC仪,配有SPD-20AT紫外检测器(日本岛津公司);XS-205型分析天平(瑞士梅特勒-托利多仪器有限公司);KQ2200B/FL0016-01超声波清洗器(昆山市超声仪器有限公司)。
1.2 药品与试剂
苯甲酸对照品(批号:100419-200301,纯度:100%)、苯甲醛对照品(批号:111650-200802,纯度:100%)均来源于中国食品药品检定研究院;甲苯对照品(美国Sigma-Aldrich公司,纯度:99.8%);药用级苯甲酸(湖南尔康制药股份有限公司,批号:20110601195、20110701195、20110801195);工业级苯甲酸(市售,批号:20110601、20110602、20110603);乙腈为色谱纯,醋酸铵、冰醋酸均为分析纯,水为重蒸水。
2 方法与结果
2.1 色谱条件
色谱柱:WondasilTM-C18(250mm×4.6mm,5μm);流动相:甲醇-0.02mol/L乙酸铵溶液(pH 4.0)(80∶20),流速:0.5ml/min;检测波长:254nm;进样量:20μl。
取“2.2”项下苯甲酸、苯甲醛、甲苯对照品溶液及系统适用性溶液进样分析,色谱见图1。
2.2 溶液的制备
2.2.1 对照品溶液的制备。分别取苯甲酸、苯甲醛、甲苯对照品适量,加流动相制备成质量浓度分别为4.0mg/ml、4.0μg/ml、3.5μg/ml的对照品溶液。
2.2.2 供试品贮备液的制备。精密称取苯甲酸原料药适量,加流动相稀释制成质量浓度约为8mg/ml的溶液,摇匀,即得。
2.2.3 系统适用性溶液的制备。即“2.2.1”项下3种对照品的混合溶液。
2.3 方法学考察
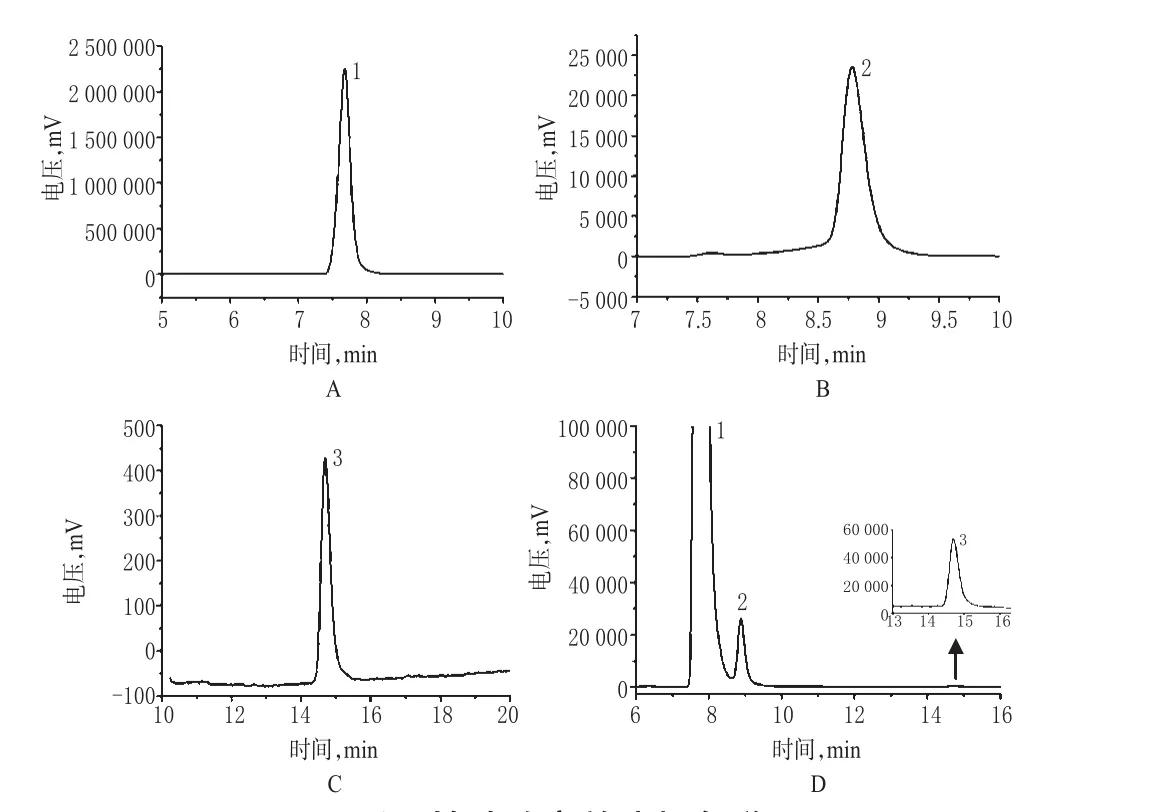
图1 适用性试验高效液相色谱图A.苯甲酸对照品溶液;B.苯甲醛对照品溶液;C.甲苯对照品溶液;D.系统适用性溶液;1.苯甲酸;2.苯甲醛;3.甲苯Fig 1 HPLC chromatograms of applicability testsA.benzoic acid control solution;B.benzaldehyde control solution;C.toluene control solution;D.system suitability solution;1.benzoic acid;2.benzaldehyde;3.toluene
2.3.1 检测波长的选择。分别取制备的苯甲酸(20μg/ml)、苯甲醛(20μg/ml)和甲苯(1.0mg/ml)对照品溶液适量,紫外扫描,结果苯甲酸、苯甲醛和甲苯最大吸收波长分别为225、245、260nm,光谱见图2。

图2 紫外吸收光谱图1.苯甲酸;2.苯甲醛;3.甲苯Fig 2 UV absorption spectrum1.benzoic acid;2.benzaldehyde;3.toluene
为灵敏检测苯甲酸有关物质,所选波长需使3种物质均有较强吸收。参考已有文献[7-9],取系统适用性溶液适量,分别选择检测波长为230、250、254nm进行试验,各物质峰面积结果见表1。

表1 不同检测波长下3种物质峰面积值结果Tab 1 Peak areas of 3kinds of substances at different detection wavelength
由表1可知,检测波长为230nm时,苯甲酸峰面积最大,苯甲醛峰面积较小,甲苯未检出;检测波长为250、254nm时,苯甲酸峰面积减小,苯甲醛、甲苯峰面积均增大。为灵敏检测苯甲醛和甲苯,故选择检测波长为254nm。
2.3.2 流动相的确定。取系统适用性溶液适量,选择甲醇与乙酸铵(0.02mol/L,pH 4.0)溶液比例分别为90∶10、80∶20、70∶30进行试验。结果,苯甲酸、苯甲醛和甲苯保留时间及分离度见表2。
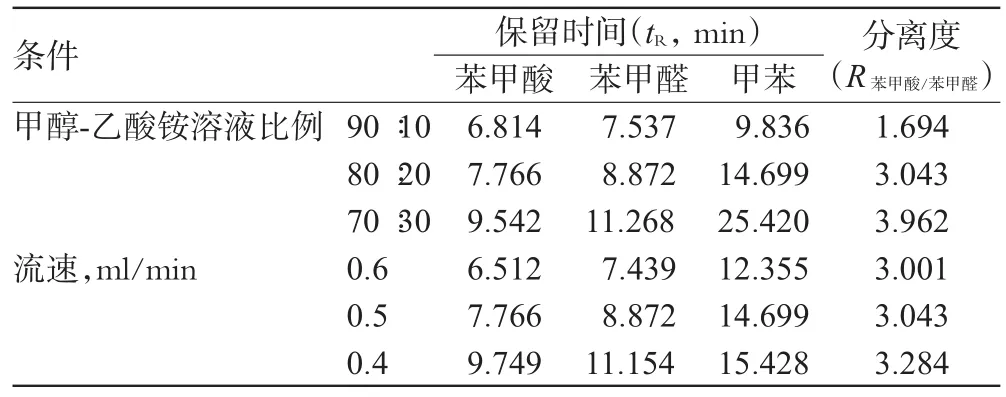
表2 不同比例流动相及不同流速下3种物质保留时间和分离度结果Tab 2 Retention time and separation rates of 3kinds of substances with different proportion of mobile phase at different flow rates
由表2可知,随着流动相中乙酸铵比例的增加,各物质保留时间随之增加,苯甲酸和苯甲醛的分离度也随之增加。当改变流动相比例从80∶20到70∶30,分离度增加不明显,但甲苯保留时间明显延长。为获得良好的分离度,并缩短色谱分析时间,选择甲醇-乙酸铵溶液(80∶20)作为流动相。
2.3.3 流速的确定。流速的大小对各物质保留时间以及分离度有较大影响,本文考察了流速分别为0.4、0.5、0.6ml/min时对保留时间及分离度的影响,结果见表2。
由表2可知,随着流速的减慢,各物质保留时间随之延长,苯甲酸和苯甲醛的分离度也随之增加。为获得良好的分离度,并缩短色谱分析时间,选择流速为0.5ml/min。
2.3.4 专属性考察。(1)未破坏溶液的配制:精密量取供试品贮备液5ml,加流动相稀释定容至10ml,备用。
(2)酸(碱)破坏性试验:精密取供试品贮备液5ml,置于试管中,加1mol/L HCl(NaOH)溶液2ml,摇匀,置于85~90℃水浴中加热4h,放冷至室温,用1mol/L NaOH(HCl)溶液中和,加流动相稀释定容至10ml,滤过,取续滤液作为酸(碱)破坏溶液。
(3)氧化破坏试验:精密取供试品贮备液5ml,置于试管中,加30%双氧水溶液2ml,摇匀,放置2h,加流动相稀释定容至10ml,滤过,取续滤液作为氧化破坏溶液。
(4)光照破坏试验:精密取供试品贮备液5ml,置于4700lx强光下照射24h,放冷至室温,加流动相稀释定容至10ml,滤过,取续滤液作为光照破坏溶液。
(5)高温破坏试验:精密取供试品贮备液5ml,置于85~90℃水浴中加热4h,放冷至室温,加流动相稀释定容至10ml,滤过,取续滤液作为高温破坏溶液。
取上述6种溶液进样分析,色谱图结果表明该色谱条件下方法专属性较好,详见图3。
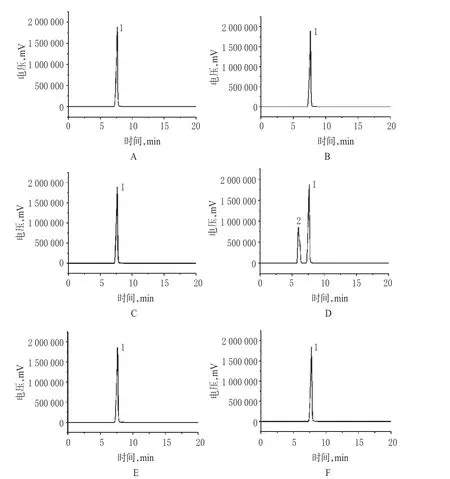
图3 破坏性试验高效液相色谱图A.未破坏溶液;B.酸破坏溶液;C.碱破坏溶液;D.氧化破坏溶液;E.光照破坏溶液;F.高温破坏溶液;1.苯甲酸;2.过氧化氢Fig 3 HPLC chromatograms of destructive testA.solution undestroyed;B.solution destroyed by acid;C.solution destroyed by alkali;D.solution destroyed by oxidation;E.solution destroyed by light;F.solution destroyed by high temperature;1.benzoic acid;2.hydrogen peroxide
2.3.5 线性关系与检测限试验。取苯甲醛、甲苯对照品制备成为质量浓度分别为4.0、3.5μg/ml的溶液,再制备成相应的50%、80%、100%、120%、180%的系列溶液,进样。以峰面积积分值(y)为纵坐标、质量浓度(x)为横坐标,得回归方程分别为苯甲醛:y=94295x-20763(r=0.9995);甲苯:y=2462.9x-482.74(r=0.9989)。结果表明,苯甲醛、甲苯检测质量浓度线性范围分别为2.0~7.2、1.75~6.30μg/ml。信噪比为3时得苯甲醛及甲苯的检测限分别为0.05、0.2μg/ml。
2.3.6 精密度试验。取“2.3.5”项下100%溶液20μl重复进样6次,记录色谱图及峰面积积分值。结果,苯甲醛、甲苯对应峰面积积分值的RSD分别为1.5%、1.3%,表明仪器精密度良好。
2.3.7 稳定性试验。分别于制备后0、2、4、6、8h取“2.3.5”项下100%溶液适量20μl,注入色谱仪,记录色谱图及峰面积积分值。结果,苯甲醛、甲苯峰面积的RSD分别为0.97%、1.02%(n=6),表明溶液在8h内基本保持稳定。
2.3.8 加样回收率试验。精密吸取供试品贮备液9份各5.0ml,分别置于25ml量瓶中,3份1组,分别添加苯甲醛及甲苯对照品溶液适量,加流动相稀释并定容至刻度,摇匀。进样,记录色谱图,计算加样回收率,结果详见表3。
2.4 苯甲醛、甲苯限度的确定
为进一步控制苯甲酸原料药的质量,对原料药中有关物质苯甲醛、甲苯进行控制,结合2010年版《中国药典》(二部)对有关物质的限度要求,建立药用级苯甲酸中有关物质苯甲醛、甲苯的测定方法,方法如下:
供试品溶液的制备:精密称取苯甲酸原料药适量,加流动相稀释制成质量浓度约为4mg/ml的溶液,摇匀,即得。

表3 回收率测定结果(n=9)Tab 3Results of recovery tests(n=9)
杂质对照溶液:分别精密称取苯甲醛、甲苯对照品适量,置于同一量瓶中,加流动相稀释制成含苯甲醛4.0、甲苯3.5μg/ml的混合溶液,摇匀,即得。
HPLC条件:色谱柱为C18;检测器为紫外检测器;流动相为甲醇-0.02mol/L乙酸铵(pH 4.0)=80∶20,流速为0.5ml/min;检测波长为254nm。
限度规定:供试品溶液中苯甲醛、甲苯的峰面积不得大于杂质对照溶液的峰面积(即苯甲醛、甲苯的限度分别为0.10%和0.0875%)。
2.5 样品中有关物质测定
分别吸取对照品溶液和供试品溶液20μl注入色谱仪,记录保留时间及峰面积积分值,外标法计算即得。对3批市售工业级及3批药用级苯甲酸样品进行了检测,结果详见表4。

表4 6批样品中苯甲醛及甲苯含量测定结果(n=3)Tab 4 Content determination results of benzaldehyde and toluene in 6batches of samples(n=3)
由表4可见,3批工业级样品中苯甲醛的含量均超出规定的限度,甲苯未检出;药用级样品中均未检出苯甲醛、甲苯,符合药用要求。
3 讨论
孙远华等[4]采用气相色谱法同时测定甲苯氧化产物中的甲苯及苯甲醛。该法需用NaOH处理样品以除去苯甲酸,再用气相色谱法分析甲苯和苯甲醛的含量,操作不便,且NaOH会影响苯甲醛的测定。阚显文等[6]采用现场电位滴定法,用NaOH标准溶液作为滴定剂同时测定苯甲醛和苯甲酸的含量。该法是基于酸碱反应的原理进行测定,专属性差,酸性物质对测定结果会产生较强的干扰。康全影等[7]采用紫外法同时检测苯甲醛、苯甲酸,其混合物在234nm波长处的吸光度值只与两者总浓度有关,250nm波长处的吸光度值与苯甲醛浓度成正比,两者相互影响较大。本文建立了苯甲酸原料药中有关物质苯甲醛、甲苯的检查方法,并规定苯甲醛、甲苯的的含量不得过0.10%和0.0875%,与已有方法相比,该方法简便、灵敏、专属性强,能同时测定3种物质,且各物质之间未见干扰,测定结果准确。此外,苯甲酸在《中国药典》标准中没有设立有关物质苯甲醛、甲苯检查项,故本法的建立有利于药品质量的有效控制。
[1] 国家药典委员会.中华人民共和国药典:二部[S].2010年版.北京:中国医药科技出版社,2010:437.
[2] 王忠元,张艳熹.甲苯液相空气氧化制苯甲酸[J].黑龙江石油化工,1996(1):1.
[3] 吴鑫干,陈舒伐.苯甲酸的合成和精制[J].现代化工,2000,20(8):10.
[4] 孙远华,张同来,张建国,等.气相色谱法测定甲苯氧化产物中的甲苯及苯甲醛[J].分析科学学报,2005,21(2):229.
[5] 刘敬兰,周鸿娟,陈连文,等.气相色谱法测定甲苯氧化产物中的苯甲醛及苯甲酸[J].色谱,1996,14(1):79.
[6] 阚显文,李茂国,陶海升,等.电位滴定法同时测定电合成产物苯甲醛和苯甲酸[J].应用化学,2003,20(7):699.
[7] 康全影,马丽,刘雄民,等.苯甲醛、苯甲酸的紫外光谱特性及含量测定方法研究[J].应用化工,2007,36(3):295.
[8] 李彦威,杨秀花,方慧文,等.电合成产物苯甲醛的高效液相色谱法测定[J].分析试验室,2009,28(1):45.
[9] 包娜,谭红,谢锋,等.高效液相色谱法测定水中的苯系物[J].贵州化工,2011,36(1):34.
[10] 孙全乐,蔡广知,贡济宇.超高效液相色谱法测定人参中人参皂苷Re的含量[J].中国药房,2012,23(3):258.
[11] 高振强,刘珠.HPLC法测定盐酸氯哌丁原料药的有关物质[J].中国药房,2008,19(22):1738.
