基于刚性配体的一种铜(Ⅱ)配合物的合成、表征、晶体结构及DFT 计算
2013-08-20王友松王萃娟张亚军邱光美周先礼王尧宇
王友松 王萃娟*, 张亚军 邱光美 黄 帅 周先礼 王尧宇
(1 西南交通大学生命科学与工程学院化学化工系,成都 610031)
(2 西北大学化学与材料学院,合成与天然功能分子化学教育部重点实验室,西安 710069)
0 Introduction
The design and construction of coordination compounds with unique structural and unique chemical and physical properties has attracted extensive interest in supramolecular chemistry and materials chemistry[1-6]. A major reason for this interest is the promise of being able to generate new materials with intriguing architectures and potential applications[7-8].Usually, the construction of coordination architecture depends on the combination of several factors[9-10], such as the coordination geometry of metal ions, the nature of organic ligands and counterions, and sometimes the different reaction conditions. For example, changes in counterions[11]and reaction conditions[12]can result in a remarkable class of materials bearing diverse architectures and functions. Thus, understanding how these considerations affect metal coordination and influence crystal packing is at the forefront of controlling coordination supramolecular arrays[13].
In the previous work[14], we reported five novel copper(Ⅱ)compounds, one nickel(Ⅱ)compound based on a rigid ligand, N,N′-di(2-pyridyl)-2,4-diamino-6-phenyl-1,3,5-triazine (Hdpdapt), and coligands. These superstructures were affected by the incorporation of different metal ions and coligands, concomitant with diverse physical properties. To extend the work about Hdpdapt, we use a special synthesis method-ultrasonic wave stir to dissolution the reagent and investigate how synthesis method affect metal coordination and influence crystal packing. Fortunately, when we introduce Hcro and Cu(OAc)2·2H2O to react with Hdpdapt under the synthesis condition as mentioned above, a new coordination compound, [Cu(dpdapt)(cro)]·CH3OH (1) was constructed. The interesting fact is that in compound 1, deprotonated dpdapt act as anion ligand to complete the metal coordination as well as compensate the charge. This is different from the compound of Hdpdapt we have reported[14]. Herein,we report the synthesis, X-ray single crystal structure of coordination compound, [Cu(dpdapt)(cro)]·CH3OH(1) together with DFT studies to explain its coordination behavior.
1 Experimental
1.1 Materials and physical measurements
The ligand Hdpdapt was prepared as we reported before[14]. All reagents and solvents employed were commercially available and used as received without further purification. Infrared spectra on KBr pellets were recorded on a Nicolet 170SX FT-IR spectrophotometer in the range 4 000 ~400 cm-1. Elemental analyses were determined with a Perkin-Elmer model 240C instrument. Thermal analyses were performed on a NETZSCH STA 449C microanalyzer with a heating rate of 10 ℃·min-1under air atmosphere. Luminescence spectra for the solid samples were recorded with a Hitachi 850 fluorescence spectrophotometer.
1.2 Synthesis of [Cu(dpdapt)(cro)]·CH3OH (1)
Complex 1 was obtained by the reaction of Cu(OAc)2·2H2O, Hdpdapt, Hcro and NaOH in molar ratio of 1∶1∶1∶2 mixed with 15 mL of aqua-methanol(Vwater/Vmethanol=1∶1)under ultrasonic wave stir conditions(at room temperature for 1 h). Diffused in Et2O after filtering and green needle-shaped crystal produced 1.(yield: 78% based on dpdapt). C24H23CuN7O3(521.03):Calcd.(%): C 55.32, H 4.45, N 18.82; Found (%): C 55.22, H 4.50, N 18.76. IR (KBr) ν: 3 300m, 1 581s,1 339s, 1 250w, 1 210w, 1 059m, 839w, 770m cm-1.
1.3 Crystal structure determination
Single-crystal X-ray diffraction of complexes 1 was performed on a BRUKER SMART 1000 CCD diffractometer equipped with a graphite crystal monochromator situated in the incident beam for data collection. Crystallographic data were collected with Mo Kα radiation(λ=0.071 073 nm)for three complexes at 273 (2) K. The structures were solved by direct methods and refined by full-matrix least squares method on F2values using SHELXL 97 and SHELXL 97 programs, respectively[15-16]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced at calculated positions. Crystal data, data collection, and refinement parameters for three complexes are shown in Table 1, and selected bond lengths and bond angles are listed in Table 2.
CCDC: 873304.
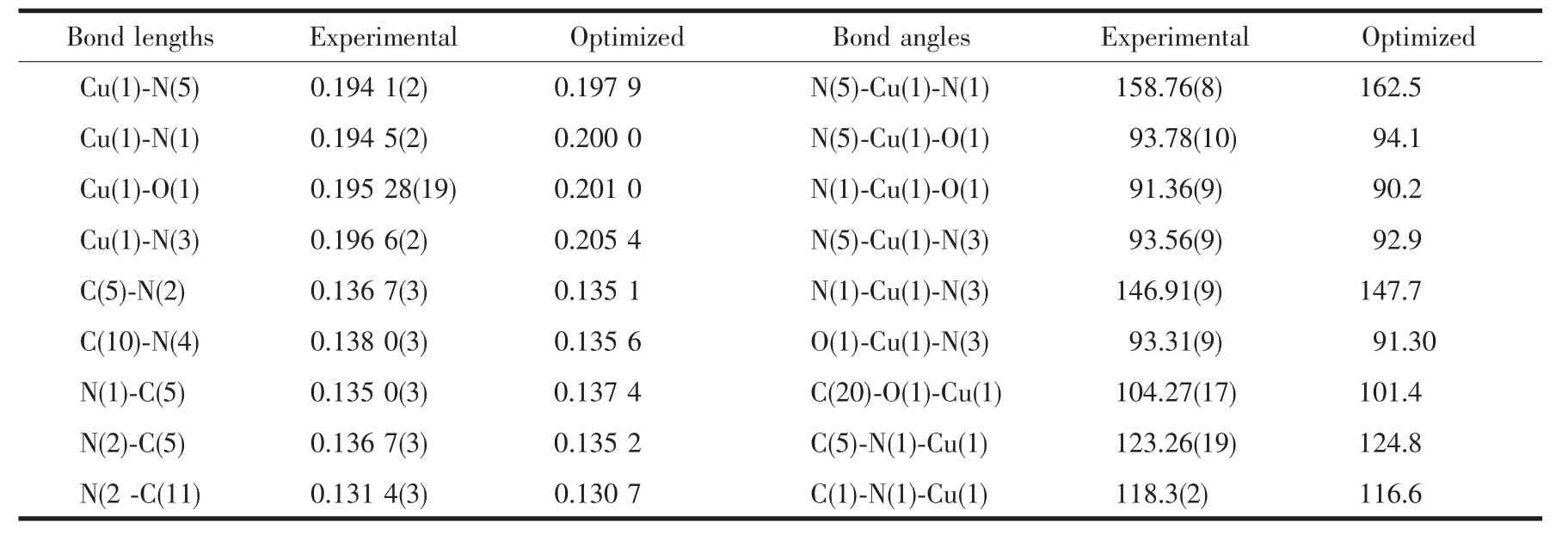
Table 2 Selected bond lengths (nm) and bond angles(°) of [Cu(dpdapt)(cro)]·CH3OH (1)
1.4 Computational details
The calculations were carried out using Gaussian03 program suit[17], including optimized geometries and calculation of vibrational frequencies were carried out at the B3LYP[18-19]level of theory.Mulliken population analysis was also performed under this method. We employed 3-21G basis set for H,C,N and,and the LANL2DZ effective core potential(ECP) set of Hay and Wadt[20]for Cu. Geometry optimization was performed for complex 1, and the attainment of energy minimum was verified by calculating the vibrational frequencies that result in absence of imaginary eigenvalues.
2 Results and discussion
2.1 Structure of [Cu(dpdapt)(cro)]·CH3OH (1)
The structure of compound 1 crystallizes in the monoclinic system form with P21/n space group, and the coordination geometry around copper(Ⅱ)center is a rare pyramid, the equatorial plane of which comprises three nitrogen atoms of dpdapt ligand; oxygen atom from one unidentate carboxylate group occupy the remaining apical coordination sites, as shown in Fig.1.Nevertheless, copper (Ⅱ)ions in coordination compounds of Hdpdapt as we reported before all sit in an five-coordinated environment with trigonal bipyramidal. Different coordination environment of metal ion is mainly origin from different synthesis methods. The Cu-O 0.195 28(19)nm and Cu-N 0.194 1(2)~0.196 6(2)nm, which are consistent with those values reported for Cu-carboxylate and Cu-pyridyl complexes[21]. The charge neutrality is achieved by the one deprotonated carboxylate group of cro molecule and one deprotonated amino-group (N2) of Hdpdapt ligand, which are very different from the Cu-Hdpdapt complexes we have reported[14].

Fig.1 ORTEP drawing of mocular structure of 1 with atom labeling scheme

Fig.2 Two-dimensional (2D) wavelike layer of 1 viewed in bc plane
The N4-H…O2 (N4-O2#1, 0.284 2(3) nm)(Symmetry codes: #1: 2-x, 1-y, 1-z) hydrogenbonding interactions between the amino-group (N4) of-dpdapt-ligand and adjacent -COO-anion (O2) lead to the formation of the antiparallel dimer motif in complex 1. Since the shortest distance from the host atom (N2) to the carboxylic oxygen atom (O3) is 0.289 1(3)nm,the guest CH3OH molecules are stabilized by hydrogen bonding interactions. And these dimer units are held together by means of face to face π…π interactions involving the triazine ring and pyridy ring(center to center distance is 0.377 7 nm and the dihedral angle is 7.15°), which further assemble into a 1D supramolecular chain along crystallographic a axis(Fig.3). Moreover, The adjacent chains are connected together via edge to face π … π interactions (the nearest distance is 0.353 4 nm) of two benzene units,and form [Cu(dpdapt)(cro)]nas wavelike layers (Fig.3).

Fig.3 3D representation of HOMO and LUMO frontal orbitals of free compound 1
2.2 Computational results
To get an insight of the electronic structures and bonding properties of the studied complex, the calculations in the DFT method were carried out. The optimized bond lengths and angles are presented in Table 2. In general, the calculated bond lengths and angles are in agreement with experimental crystal data and the large differences (~0.020 nm, ~3°) may be noticed for deviations around the center copper and N2 atom.
Mulliken charge of the center metal Cu (Table 3)change from +2 to 0.472, it′s obviously Cu has been coordinated by the ligand and its charge transfer to the ligand. The net atomic charge of coordinated N(1)N(3), N(5) and O(1) atoms are negative. Compared to the free Hdpdapt ligand (N(5), -0.535; N(1) and N(3),-0.556; N(6) and N(7), -0.618), the change about charge distribution value of donor nitrogen atoms(N(1), N(3), N(5)) are more than that of the noncoordinated nitrogen atoms (N(6), N(7)) in triazine. All these show strong complexation between Cu and its coordinated atoms. It′s worth to note that the net atomic charge of deprotonated N2 is -0.661 and undeprotonated N4 is -0.863, whereas, in free Hdpdapt ligand, the net atomic charge of N2 and N4 is also -0.863. The difference net atomic charge of N2 and N4 in compound 1 confirms that different environment of N2 and N4 in compound 1.

Table 3 Mulliken atomic charges (e) for 1 and ligand
Based on the natural charges and electron configurations on the atoms of complex 1 and free Hdpdapt ligand which have been calculated by natural bond orbital (NBO) analysis (Table 4), one can find out that, the natural charge of coordinated nitrogen atoms have been increased to -0.594, -0.574 and -0.696 in compound 1, from -0.427, -0.427 and-0.503 respectively for free Hdpdapt ligand. These results verify that in the complex formation process,the Cu atom electron density may be increased. So the electron density shifts from copper to into the coordinated nitrogen atoms. It is clear, this case causes to increasing the nature charge of coordinated nitrogen atoms and decreasing the bond order of N1-C5 bond (optimized: 0.1374 nm, experiment: 0.135 0(3) nm). Also the natural charge of C5 and C11 has been decreased to 0.206 and 0.308 in the complex 1,from 0.386 and 0.616 for the free ligand. The notable fall of natural charge of C5 and C11 result from: the electron density shift from C5 and C11 into the deprotonated N2 atom. Thus, the bond order of N2-C5 and N2-C11 has been increased to 0.135 2 and 0.130 7 nm, from 0.136 6(3) and 0.136 0(3) nm respectively. All of this testified that the synergistic effect have been occurred in the formation process of compound 1.
Schematic representation of HOMO and LUMO orbitals for 1 is presented in Fig.3. The copper dπatomic orbital (dxz, dyzand dxy) and π orbitals of N2 atom make the main contributions into the HOMO-1 and HOMO. The LUMO orbital has predominantly π*(dpdapt) character. The results approved its coordination behavior and the value of ΔE (ΔE=ELUMO-EHOMO) is 3.47 eV, which shows that the complex 1 can exist stably.
2.3 TGA Analyses

Table 4 Natural configurations and natural charges for the atoms of the compound 1 which calculated by B3LYP method
The existence of the guest molecule inspired us to investigate the thermal stability of the framework.Thermogravimetic anslyses (TGA) showed that 1 lost 5.8% of total weight in the 60 ~100 ℃temperature range, corresponding to the loss of one methanol molecule per formula unit (Calcd. 6.1%). When the temperature is above 320 ℃, the product begins to decompose and oxidize. The residual percentage weight (Obsd. 13.6%) at the end of the decomposition of the complex is consistent with the formation of CuO(Calcd. 15.2%).
3 Conclusions
In summary, we have developed a new strategy for constructing [Cu(dpdapt)(cro)]·CH3OH (1). In compound 1, deprotonated dpdapt act as anion ligand to complete the metal coordination as well as compensate the charge. Compound 1 exhibits 1D supramolecular chain formed by π-π stacking interactions, featuring antiparallel dimer motif. The DFT calculations of the complex 1 and free Hdpdapt ligand at the B3LYP level of theory verifies that the special coordination behavior have been occurred in the compound 1. Our results provide an effective and controllable route for the preparation of supramolecular architectures.
[1] (a)Hoskins B F,Robson R.J.Am.Chem.Soc.,1990,112:1546-1549
(b)Yaghi O M, Li H, Davis C, et al. Acc. Chem. Res., 1998,31:474-482
[2] (a)Yaghi O M,O′Keeffe M,Ockwig N W,et al. Nature, 2003,423:705-714
(b)Batten S R, Robson R. Angew. Chem., Int. Ed. Engl.,1998,37:1460-1494
[3] (a)Moulton B, Zaworotko M J. Chem. Rev., 2001,101:1629-1658
(b)Wang C J, Ma H R, Wang Y Y, et al. Cryst. Growth Des.,2007,7:1811-1817
[4] DING Yu-Jie(丁玉洁), WANG Yang(王扬). Chinese J. Inorg.Chem.(Wuji Huaxue Xuebao), 2011,7(27):1411-1416
[5] Li D S, Fu F, Zhao J, et al. Dalton Trans., 2010,39:11522-11525
[6] Pan P B, Sun C F, Chen S M, et al. Inorg. Chem. Commun.,2011,14:1333-1336
[7] Bardají M, Barrio M, Espinet P. Dalton Trans., 2011,40:2570-2577
[8] YANG Ying-Qun(杨颖群), CHEN Man-Sheng(陈满生),CHEN Zhi-Min(陈志敏),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao), 2011,9(27):1847-1851
[9] Wang C J, Wang Y Y, Liu J Q, et al. Inorg. Chim. Acta,2009,362:543-550
[10]Wibowo A C, Smith M D, zur Loye H C. Cryst. Growth.Des., 2011,11:4449-4457
[11](a)Vreshch V, Shen W T, Nohra B, et al. Chem. Eur. J.,2012,18:466-477
(b)Kounavi K A, Manos M J, Moushi E E, et al.CrystEngComm, 2012,12:429-438
[12](a)Peresypkina E V, Vostrikova K E. Dalton Trans., 2012,41:4100-4106
(b)Wriedt M, Zhou H C. J. Dalton Trans., 2012,41:4207-4216
[13](a)Knope K E, Kimura H, Yasaka Y, et al. Inorg. Chem.,2012,51:3883-3890
(b)Jess I, Boeckmann J, Nather C. Dalton Trans., 2012,41:228-236
[14](a)Wang C J, Ma H R, Wang Y Y, et al. Cryst. Growth. Des.,2007,7:1811-1817
(b)Wu Y P, Wang C J, Wang Y Y, et al. Polyhedron, 2006,25:3533-3542
(c)Wang C J, Ren P D, Zhang Z B, et al. J. Coord. Chem.,2009,62:2814-2823
[15]Sheldrick G M. SHELXL-97, Program for Crystal Structure Determination, University of Göttingen, Germany, 1997.
[16]Sheldrick G M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
[17]Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03.Revision B.03, Gaussian, Inc., Pittsburgh, PA, 2003.
[18]Becke A D. J. Chem. Phys., 1993,98:5648-5652
[19]Lee C, Yang W, Parr R G. Phys. Rev., 1988,37:785-789
[20]Vrkic A K, Taverner T. Dalton Trans., 2004:197-208
[21](a)Mahmoudkhani A H, Shimizu G K H. Inorg. Chem., 2007,46:1593-1602
(b)Ma L F, Wang L Y, Huo X K, et al. Cryst. Growth Des.,2008,8:620-628
(c)Liu J Q, Wu W P, Wang Y Y, et al. Inorg. Chem. Acta,2009,362:1295-1301
(d)Zhou C H, Wang Y Y, Sheng D S, et al. Eur. J. Inorg.Chem., 2006,18:2437-2446
