二茂铁取代卟啉的合成表征及电化学性质
2013-10-17朱佳丽张秀风房媛媛路桂芬欧忠平
朱佳丽 张秀风 房媛媛 孙 斌 路桂芬 欧忠平
(江苏大学化学化工学院,镇江 212013)
0 引 言
卟啉(简写为P)及其衍生物是一类由4个吡咯环通过亚甲基连接而成的具有大共轭结构的芳香性大环化合物,它们结构稳定并具有供电子和吸电子的双重功能,因而在功能材料、生物医药、光催化和电催化剂、电化学传感器等领域都有着广泛的应用前景[1-8]。二茂铁是由两个环戊二烯和二价铁形成的夹心型非苯系芳香族化合物。将二茂铁引入卟啉分子可以形成许多具有独特的电子和空间结构、特殊的光谱和电化学性质的化合物,因此该类化合物的合成及性质研究已引起有关领域研究工作者的关注[9-17]。
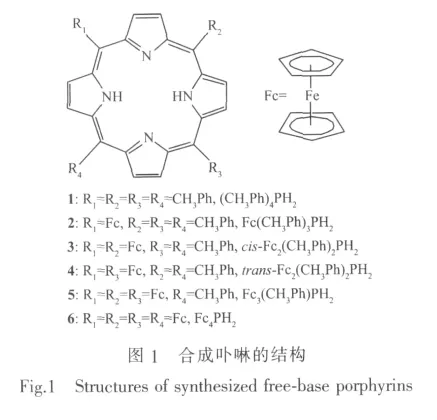
本文在二氯甲烷溶液中用吡咯与对甲基苯甲醛和二茂铁甲醛进行交叉缩合反应,合成并成功分离纯化了一系列共6个分别含有0~4个二茂铁取代基的卟啉化合物(见图1),其中4个化合物(2~5)未见文献报道。借助紫外-可见光谱、红外光谱、核磁共振、质谱等技术对所合成的卟啉化合物进行了表征,测定了每个化合物在非水溶剂中的质子化反应常数,研究了它们的电化学和光谱电化学性质,讨论了二茂铁取代基对化合物的光谱及氧化还原电位的影响。
1 实验部分
1.1 试剂与仪器
二氯甲烷(CH2Cl2)、正己烷和 N,N′-二甲基甲酰胺(DMF)的提纯:取一定量分析纯试剂置于250 mL磨口烧瓶中,用无水MgSO4干燥24 h,抽滤,滤液转移至250 mL烧瓶中,加入适量的P2O5后蒸馏,收集中间馏分备用。二氯甲烷与正己烷采用常压蒸馏,DMF采用减压蒸馏。
四正丁基高氯酸铵(TBAP):取100 g粗品TBAP放入250 mL烧杯中,加无水乙醇100 mL,加热溶解后趁热抽滤。将滤液转移至原烧杯中置于冰箱中冷却,结晶析出后抽滤、用30 mL无水乙醇分2~3次洗涤,将洗涤后的晶体转移至烧杯中置于70~80℃的烘箱中干燥5~7 d,烘干后的TBAP置于棕色广口瓶中保存备用。
8453安捷伦紫外可见分光光度计;Nexus470傅里叶红外光谱仪;Bruker AvancⅡ400 MHz核磁共振仪;Bruker BIFLEXⅢ型质谱仪;VARIAN Cary Eclipse荧光分光光度计;CHI730C电化学工作站。
1.2 实验方法
电化学测定采用三电极系统,工作电极为玻碳电极(GCE),辅助电极为铂丝电极,参比电极为饱和甘汞电极(SCE),其中SCE与待测溶液之间用盐桥隔离。电位扫描前溶液通氮气除氧,扫描过程保持在N2条件下进行。原位紫外可见光谱电化学测定在具有二极管矩阵阵列检测器的8453安捷伦分光光度计上进行,用CHI-730C电化学工作站控制电位。自制的薄层光谱电化学池采用三电极体系,其中铂网栅为工作电极,铂丝为辅助电极,饱和甘汞电极为参比电极。用盐桥隔开参比电极与整体溶液,实验在N2保护下进行。
微量光谱滴定法测定卟啉的质子化反应常数:以二氯甲烷作溶剂用逐级稀释法配制一系列不同浓度的三氟乙酸(HA)标准溶液。用微量注射器将一定浓度的HA溶液逐渐定量加入到含有卟啉化合物溶液的光谱滴定池中,摇匀后在分光光度计上测定并记录相应的光谱。当加入HA溶液时光谱不再有明显变化时完成滴定。
无金属卟啉分子中的两个内核N在酸性溶液中可以接受质子生成质子化产物,根据下面的Hill方程[18-19]可以求得质子化反应常数lgβn。
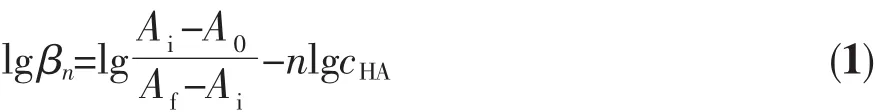
式中,A0为完全没有质子化时溶液的吸光度,Ai为滴定过程中任意一点时溶液的吸光度,Af为卟啉完全质子化后溶液的吸光度。根据一定波长下的吸光度,由lg[(Ai-A0)/(Af-Ai)]对lgcHA作图,得到的曲线斜率即为化合物接受质子的数目n,而由截距可求得质子化反应常数lgβn。
1.3 合成方法
二茂铁取代卟啉化合物的合成路线见Scheme 1。
在盛有150 mL二氯甲烷的三颈瓶中,加入二茂铁甲醛 0.60 g,对甲基苯甲醛 0.30 g(物质的量之比为 1∶1),新蒸吡咯 0.40 g(两种醛的总量与吡咯的物质的量之比约为1∶2)和三氟化硼乙醚0.05 mL,于室温氮气保护下搅拌反应20 h后分4次加入四氯苯醌 0.98 g,然后加热回流 3.5 h。 反应过程中用TLC检测反应进度。反应结束后将溶液蒸发浓缩。浓缩液先用碱性氧化铝色谱柱进行粗分 (洗脱剂为V二氯甲烷∶V正已烷=1∶1),分别收集 6 个不同色带的溶液。然后用硅胶柱对每个色带的溶液作进一步的纯化分离,所用的洗脱剂根据不同色带的溶液分别采用1∶2,1∶1,3∶2,3∶1 或 1∶0 的二氯甲烷和正己烷的混合溶剂。收集洗脱液,在80℃下旋转蒸发得到紫色粉末状固体后在二氯甲烷和正己烷的混合溶剂(1∶100)中进行重结晶,即得相应的纯化合物。
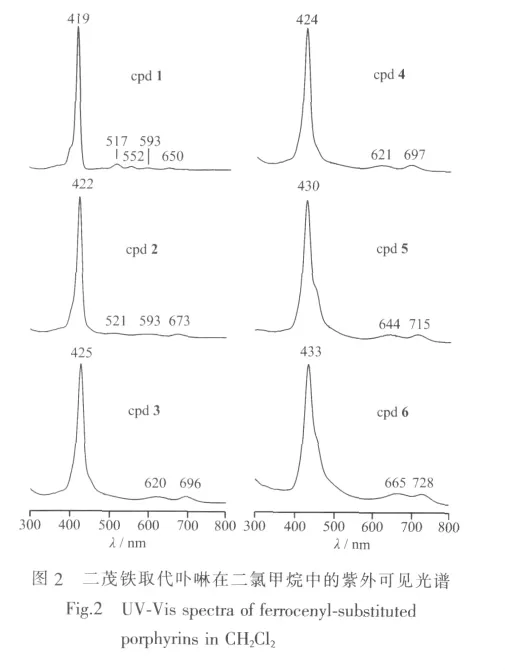
化合物 1(CH3Ph)4PH2,23 mg,产率 2.8%;UVVis(λmax/nm,CH2Cl2):419,517,552,593,650;IR(KBr,cm-1)ν:3449,3307,3024,2915,1506,1471,1219,1153,1182,966;1H NMR(CDCl3,δ/ppm):8.88(s,8H,β-pyrr),8.11~8.13(d,8H,o-Ph),7.57~7.59(d,8H,m-Ph),2.73(s,12H,Me-H),-2.74(s,2H,NH);MS(MALDI-TOF):(M)670.98,Calcd.670.84。
化合物 2 Fc(CH3Ph)3PH2,127 mg,产率 13.1%;UV-Vis(λmax/nm,CH2Cl2):422,521,593,673;IR(KBr,cm-1)ν:3442,3311,3088,3021,2922,2853,2358,1 557,1 508,1 470,1 223,1 184,1 153,967;1H NMR(CDCl3,δ/ppm):10.00(s,2H,β-pyrr-Ph-Fc),8.79 ~8.82(t,6H,β-pyrr-Ph-Fc),8.12(s,6H,o-Ph),7.58(s,6H,m-Ph),5.58(s,2H,α-Cp),4.85(s,2H,β-Cp),4.20(s,5H,CpH),2.72~2.74(d,9H,Me-H),-2.29(s,2H,NH);MS(MALDI-TOF):[M+H]+766.10,Calcd.764.74。
化合物 3 cis-Fc2(CH3Ph)2PH2,14 mg,产率 1.9%;UV-Vis(λmax/nm,CH2Cl2):425,620,696;IR(KBr,cm-1)ν:3 294,2 958,2 921,2 852,2 359,1 556,1 508,1 467,1 225,1 183,1 158,968;1H NMR(CDCl3,δ/ppm):9.83~9.94(d,4H,β-pyrr-Fc),8.71~8.77(d,4H,β-pyrr-Ph),8.08~8.10(d,4H,o-Ph),7.58(s,4H,m-Ph),5.52(s,4H,α-Cp),4.84(s,4H,β-Cp),4.14(s,10H,CpH),2.73(s,6H,Me-H),-1.78(s,2H,NH);MS(MALDI-TOF): [M+H]+860.31,Calcd.858.63。
化合物 4 trans-Fc2(CH3Ph)2PH2,5 mg,产率 0.67%;UV-Vis(CH2Cl2,λmax/nm):424,621,697;IR(KBr,cm-1)ν:3448,3300,3092,3021,2919,2359,1557,1508,1472,1229,1182,1153,968;1H NMR(CDCl3,δ/ppm):9.83(s,4H,β-pyrr-Fc),8.707(s,4H,β-pyrr-Ph),8.705(s,4H,o-Ph),7.75(s,4H,m-Ph),5.526(s,4H,α-Cp),4.837(s,4H,β-Cp),4.141(s,10H,CpH),2.737(s,6H,Me-H),-1.66(s,2H,NH);MS(MALDI-TOF):[M+H]+860.31,Calcd.858.63。
化合物 5(Fc)3(CH3Ph)PH2,37 mg,产率 4.7%;UV-Vis(λmax/nm,CH2Cl2):430,644,715;IR(KBr,cm-1)ν:3 449,3 300,3 090,2 923,2 853,2 361,1 557,1 509,1469,1251,1187,1155,969;1H NMR(CDCl3,δ/ppm):9.79(s,4H,β-pyrr-Fc-Fc),9.69(s,2H,β-pyrr-Fc-Ph),8.67(s,2H,β-pyrr-Fc-Ph),8.07~8.09(d,2H,o-Ph),7.59(s,2H,m-Ph),5.40(s,6H,α-Cp),4.75~4.77(d,6H,β-Cp),4.02(s,15H,CpH),2.73(s,3H,Me-H),-1.05(s,2H,NH);MS(MALDI-TOF):[M+H]+954.41,Calcd.952.52。
化合物 6(Fc)4PH2,9 mg,产率 1.23%;UV-Vis(λmax/nm,CH2Cl2):433,665,728;IR (KBr,cm-1)ν:3 449,3 300,3 090,2 923,2 851,2 359,1 553,1 470,1 234,1 155,971;1H NMR(CDCl3,δ/ppm):9.64(s,8H,β-Pyrr),5.36(t,8H,α-Cp),4.80(t,8H,β-Cp),4.00(s,20H,Cp-H),-0.47(s,2H,NH);MS(MALDI-TOF,m/z): (M)1 046.53,Calcd.1 046.41。
2 结果与讨论
2.1 1H NMR及质谱分析
以四甲基硅烷为内标,氘代氯仿为溶剂,测定了每个化合物的核磁共振氢谱,所得数据见实验部分。通过比较化合物1~6的核磁共振数据可以得知,与无二茂铁取代的卟啉化合物1相比,化合物2~6因为具有供电子效应的二茂铁基,因此分子中的β-氢和2个内核N-氢的化学位移(δ)均朝着低场移动。分子中二茂铁基的数目愈多,内核N-氢的δ向低场移动的愈大,但二茂铁基的数目对β-氢的δ影响较小。由化合物的质谱测定结果可知,各个卟啉的分子量测定值与理论计算基本相符,说明得到的为所期望合成的卟啉化合物。
2.2 红外光谱分析
采用KBr压片法测定了每个化合物的红外光谱。 以 5-二茂铁基-10,15,20-三(4-甲基苯基)卟啉 2为例,各主要吸收峰归属如下:3 442 cm-l处的吸收峰是卟啉分子中环内N-H键的伸缩振动峰,967 cm-1的吸收峰则是吡咯环上N-H键的弯曲振动峰,据此可以确定产物中存在N-H结构。2 922 cm-l处的吸收峰是甲基的C-H伸缩振动峰;3 088和3 311 cm-1处的吸收峰是苯环和吡咯环上的C-H伸缩振动峰;在1 508、1 470和1 153 cm-1的是对应于苯环和吡咯环的骨架振动吸收峰;1 223 cm-1的是CN键的伸缩振动吸收峰;1 184 cm-1的是苯环上CH的弯曲振动吸收峰;2 853、2 359和1 557 cm-1的是二茂铁茂环上C-H键的伸缩振动和弯曲振动吸收峰。
2.3 紫外可见光谱分析
分别测定了化合物1~6在二氯甲烷溶剂中的紫外可见光谱(见图2)。从图2可见,化合物1~6都具有1个强Soret吸收峰和2~4个弱的Q吸收峰。随着卟啉分子中二茂铁取代基(Fc)数目的增加,其Soret吸收峰由化合物1的419 nm逐渐红移至化合物6的433 nm,即每增加1个Fc取代基可以使化合物的Soret吸收峰平均红移3~4 nm。另外,二茂铁取代基的引入不仅可以使Q吸收峰发生较大的红移,而且还导致Q吸收峰的数目发生了变化。例如,没有Fc取代基的化合物1有4个弱的Q吸收峰,而含有1个Fc的化合物2有3个Q吸收峰。当化合物含有2个以上Fc取代基时(化合物3~6),在光谱上就只能观察到2个Q吸收峰。结果表明,在卟啉分子中引入具有给电子效应的二茂铁取代基,可以使分子中共轭大环上的电子云密度增大、降低电子跃迁的激发能,从而导致卟啉化合物的光谱峰发生红移。需要说明的是,化合物3和4为含有2个Fc取代基的顺反异构体,它们具有非常相似的紫外可见光谱,说明Fc取代基的位置对卟啉化合物的光谱没有显著的影响。
2.4 质子化常数的测定
在二氯甲烷溶液中,当用三氟乙酸(HA)溶液滴定卟啉化合物时,其Soret吸收峰强度会逐渐降低直至消失,但同时会在长波方向出现1个光谱强度相当的Soret吸收峰和1个较强的Q吸收峰 (见表1)。图3为化合物1、3和5在滴定过程中的光谱变化情况。利用一定波长下的吸光度并根据方程式(1)作lg[(Ai-A0)/(Af-Ai)]对lgcHA的曲线,求得卟啉接受质子的数目和质子化反应常数列于表1。


表1 质子化卟啉的紫外可见光谱数据以及化合物在298 K的二氯甲烷中测定的质子化数和平衡常数Table 1 UV-Vis spectral data(λmax,nm)of protonated porphyrins and the number(n)and equilibrium constants(lgβ2)of protonation of the compounds in CH2Cl2at 298 K
实验结果表明,在CH2Cl2中所有测定的卟啉化合物与三氟乙酸反应时均能够一步得到2个质子(n=2)生成相应的质子化产物(见表1),但是它们的质子化常数具有较大的区别。 由表1可见,化合物1的 lgβ2=8.0 而化合物 2~6 的 lgβ2要大得多,为 9.9~10.7,这说明在卟啉分子中引入具有给电子作用的二茂铁取代基,可以使其更容易被质子化。另外,由于化合物2~6的质子化常数差别不大,因此可以推断,当卟啉化合物分子中的meso位上具有1~4个Fc取代基时,对化合物的质子化反应影响不大。
2.5 电化学和光谱电化学
利用循环伏安法研究了6种无金属卟啉化合物在 CH2Cl2溶剂中存在 0.1 mol·L-1TBAP 支持电解质时的电化学行为,图4是化合物2和5的循环伏安图,表2则列出了6个化合物在扫描速率为0.10 V·s-1时的氧化还原电位。

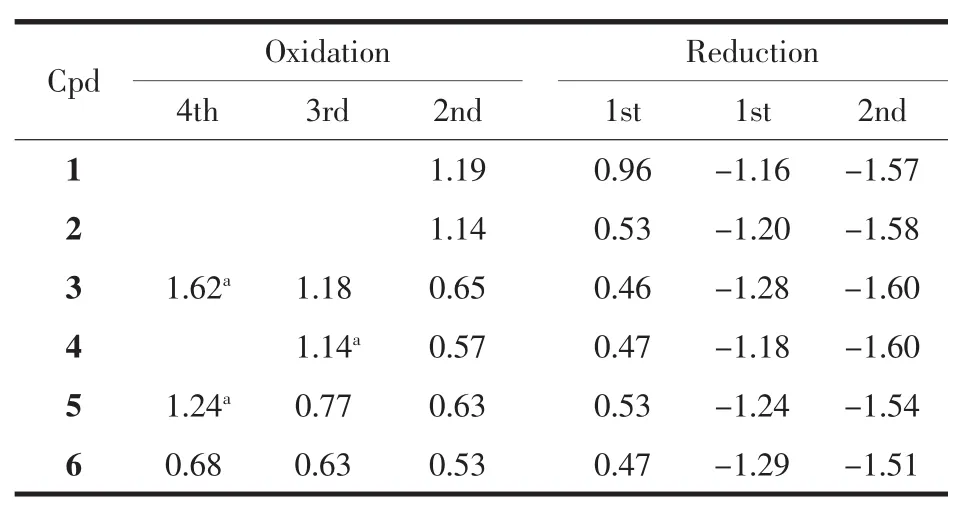
表2 二茂铁取代卟啉在二氯甲烷中的氧化还原电位Table 2 Redox potentials(V vs SCE)of free-base ferrocenylporphyrins in CH2Cl2
由表2可知,在CH2Cl2中每个化合物均能观察到两步可逆的或准可逆的还原反应。无二茂铁取代基的化合物 1,其还原电位发生在-1.16和-1.57 V,而化合物2~6含有1~4个具有给电子效应的二茂铁基,所以它们的第一步还原电位与化合物1相比均有不同程度的负移,但化合物1~6的第二步还原电位只有较小的差别。在CH2Cl2中,化合物1有两步可逆的氧化反应分别位于0.96和1.19 V,这与该化合物在相同实验条件下的文献电位值 (0.94和1.18 V)基本相同[20-21],而化合物 2~6 则可以观察到2~4步氧化反应。例如,化合物2有两步氧化反应位于 0.53 和 1.14 V(图 3a), 化合物 5 有四步氧化反应,分别位于 0.53,0.63,0.77 和 1.24 V(图 3b)。 和化合物1相比,化合物2的第一步氧化反应负移了0.43 V,而且其电位与二茂铁在相同溶剂中的氧化电位(0.49 V)非常接近(见图 3c),因此可以确定化合物2的第一步氧化反应是分子中二茂铁基团上基于Feギ/Feバ的电子转移过程。而化合物5的前3步氧化则分别为分子中3个二茂铁基团中的Feギ失去电子生成Feバ的电子转移过程。
利用光谱电化学方法可以进一步确定电极反应的产物以及电子转移机理,本文利用该方法测定了所合成的卟啉化合物在CH2Cl2溶剂中的光谱电化学性质。实验结果表明,化合物1~6在控制一定的电位还原时均可以分别生成相应的卟啉阴离子自由基和-2价阴离子。但是在控制一定的电位氧化时,没有Fc取代基的化合物1和含有1~4个Fc取代基的化合物2~6相比,生成的电极反应产物不同,其氧化反应机理也不同。
已知许多无金属的卟啉化合物在二氯甲烷溶剂中的电氧化过程(E)往往会伴随着某些化学反应(C),因此最终得到的产物并不仅仅是化合物的阳离子自由基,而且还有其质子化的产物[22]。化合物1在进行第一步控制电位电解时,也经历了同样的EC机理过程。从图5a可以看到,当控制电位1.00 V电解时,随着位于419nm波长处的Soret吸收峰强度不断降低的同时,在445和665 nm处有2个新峰生成,而这2个新的吸收峰与化合物1的质子化产物[(CH3Ph)4PH4]2+在442和668 nm波长处的最大吸收非常相似(见表1),结果表明化合物1在第一步氧化时经过EC机理生成了相同的质子化产物。
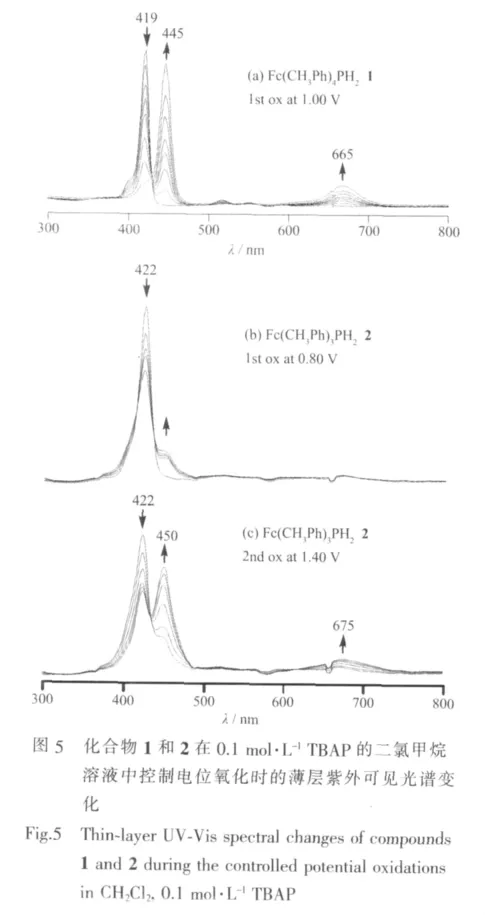
对于化合物2~6来说,控制电位电解时,是分子中的Fc取代基而不是卟啉大环首先被氧化,因此化合物Soret吸收峰强度的降低程度较小。例如,化合物2在光谱电化学池中于0.80 V电位下进行第一步氧化时,其在422 nm波长处的Soret吸收峰强度没有很大的下降(图5b),也没有强度较大的新吸收峰生成,结果说明该化合物的第一步氧化主要发生在Fc取代基上,生成的是含有Feバ的卟啉化合物,这与该化合物的循环伏安测定结果完全一致(见图 3a)。
如果在第一步氧化的基础上,对化合物2在1.40 V电位下作进一步的控制电位氧化,可以观察到在422 nm处的Soret吸收峰强度继续降低,同时在675 nm波长处逐渐生成了1个弱的宽峰(图5c),这是卟啉大环失去1个电子生成阳离子自由基时的特征光谱变化[23]。另外,随着电解的进行,化合物2在450 nm波长处还有1个较强的Soret吸收峰生成,而且其波长与该化合物在接受2个质子后生成的二价阳离子[Fc(CH3Ph)3PH4]2+所具有的波长(446 nm)十分相近(见表1)。结果表明在CH2Cl2中,化合物2的第二步氧化过程(E)伴随着一定程度的化学反应(C),因此在卟啉大环失去电子生成阳离子自由基时也有部分质子化产物生成。
3 结 论
在卟啉分子中引入具有给电子作用的二茂铁取代基,可以增大卟啉共轭大环上的电子云密度、降低电子跃迁的激发能,导致化合物的紫外可见光谱吸收峰产生红移,并使化合物更容易发生质子化反应。卟啉分子中的二茂铁取代基比卟啉大环容易氧化,如果具有多个二茂铁基团,则它们可以被分步氧化,逐步生成含有多个Feバ的卟啉化合物。
[1]Chou J H,Nalwa H S,Kosal M E,et al.The Porphyrin Handbook,Kadish K M,Smith K M,Guilard R,Ed.;New York:Academic Press,2000,Vol.6 Ch.41,pp43-132
[2]Aida T,Inoue S.The Porphyrin Handbook.Kadish K M,Smith K M,Guilard R,Ed.;New York:Academic Press,2000,Vol.6 Ch.41,pp133-156
[3]Pandey R K,Zheng G.The Porphyrin Handbook,Kadish K M,Smith K M,Guilard,R.,Ed.;New York:Academic Press,2000,Vol.6 Ch.41,pp157-230
[4]Malinski T.The Porphyrin Handbook,Kadish K M,Smith K M,Guilard,R.,Ed.;New York:Academic Press,2000,Vol.6 Ch.41,pp231-256
[5]Ogoshi H,Mizutani T,Hayashi T.The Porphyrin Handbook,Kadish K M,Smith K M,Guilard,R.,Ed.;New York:Academic Press,2000,Vol.6 Ch.41,pp279-340
[6]LUO Xian(罗贤),LÜ Gui-Qin(吕桂琴).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27:1705-1708
[7]YAN Ya(严亚),LÜ Ying(吕英),XIA Yi(夏怡),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27:1999-2004
[8]LUO Yun(罗云),SHI Yong-Ping(史永平),YAO Gui-Ping(姚桂平),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28:1139-1144
[9]Nemykin V N,Galloni P,Floris B,et al.Dalton Trans.,2008:4233-4235
[10]Nemykin V N,Rohde G T,Barrett C D.et al.Inorg.Chem.,2010,49:7497-7509
[11]Rohde G T,Sabin J R,Barrett C D,et al.New J.Chem.,2011,35:1440-1448
[12]Solntseva P V,Neisena B D,Sabina J R,et al.J.Porphyrins Phthalocyanines,2011,15:612-621
[13]Shetti V S,Ravikanth M.Eur.J.Org.Chem.,2010:494-508
[14]Kalita D,Morisue M,Kobuke Y.New J.Chem.,2006,30:77-92
[15]Mansour H,El-Khouly M E,Shaban S Y,et al.J.Porphyrins Phthalocyanines,2007,11:719-728
[16]D′Souza F,Smith P M,Gadde S,et al.J.Phys.Chem.B,2004,108:11333-11343
[17]Lakshmi V,Santosh G,Ravikanth M.J.Organomet.Chem.,2011,696:925-931
[18]Ellis P E,Linard J E,Szymanski T,et al.J.Am.Chem.Soc.,1980,102:1889-1896
[19]Ou Z P,Shen J,Shao J G,et al.Inorg.Chem.,2007,46:2775-2786
[20]Kadish K M,Royal G,Van Caemelbecke E,et al.The Porphyrin Handbook,Kadish K M,Smith K M,Guilard R,Ed.;New York:Academic Press,2000,Vol.9,Ch.59,pp1-220
[21]Hariprasad G,Dahal S,Maiya B G.J.Chem.Soc.,Dalton Trans.,1996:3429-3436
[22]Inisan C,Saillard J Y,Guilard R,et al.New J.Chem.,1998,22:823-830
[23]Kadish K M,Shao J G,Ou Z P,et al.Inorg.Chem.,2005,44:9023-9038
