马铃薯膳食纤维的表征及物性分析
2013-08-07张艳荣魏春光崔海月
张艳荣,魏春光,崔海月,陈 雪
(吉林农业大学食品科学与工程学院,吉林 长春 130118)
膳食纤维通常是指由可食性植物细胞壁残余物及与之相缔合的物质构成的在人体的小肠中难以消化吸收的化合物[1],其主要包括植物性木质素、纤维素、半纤维素、果胶及动物性壳质、胶原等。继蛋白质、糖、脂肪、矿物质、水和维生素之后被列为人体必需的“第七营养素”[2]。研究表明膳食纤维可以降低冠心病的发病率、降低血清胆固醇水平[3-4]。膳食纤维包括不溶性膳食纤维和可溶性膳食纤维两大类[5],其中可溶性膳食纤维具有较强的生理功能,而大多数天然膳食纤维其可溶性膳食纤维所占比例较小。但也有报道称在对有害物质的清除能力和调节肠道功能方面,不溶性膳食纤维的作用优于可溶性膳食纤维[6]。目前膳食纤维的分析方法包括酶-质量法、酶-化学法、红外光谱技术、尺寸排阻液相色谱法和高效阴离子交换色谱法等,其中红外光谱技术具有方便、快捷、准确、高效、不破坏样品、节约能源、无污染、低成本等优点,在国内外已得到广泛应用。如:陈雪峰等[7]采用红外分析和电镜扫描对水溶性膳食纤维化学组成和表面微观结构进行了研究。潘利华等[8]采用扫描电镜和红外光谱仪等对超声辅助、酸溶碱沉、酶解辅助提取所获得的水不溶性大豆膳食纤维的化学组成和表面微观结构进行了研究。
马铃薯渣是马铃薯淀粉生产的副产物,其含水量在90%左右。由于马铃薯渣产量大、不易运输和贮存、易腐败变质产生恶臭,对环境造成严重的污染。马铃薯渣总膳食纤维含量高达80%左右,是极好的膳食纤维资源。在国内,马铃薯渣通常被用做饲料或直接废弃,综合利用程度较低。国外有将马铃薯渣添加到食品中作为纤维添加剂和脂肪替代物等[9]。目前国内有关马铃薯膳食纤维结构成分的研究报道很少,吕金顺等[10]研究了水蒸汽爆破和氧化法对马铃薯膳食纤维结构的影响,有关马铃薯膳食纤维平均相对分子质量、聚合度的报导尚未见到。本研究以马铃薯渣为原料,制备马铃薯膳食纤维,对其平均相对分子质量、聚合度及物性进行测定,并利用红外光谱技术分别对马铃薯膳食纤维中可溶性与不溶性膳食纤维的官能团结构和组成进行分析,为马铃薯膳食纤维功能研究及在食品工业中应用提供理论依据。
1 材料与方法
1.1 材料与试剂
无霉变、无腐烂新鲜马铃薯 市售;耐高温α-淀粉酶(酶活力4×104U/g)、Alcalase 2.4L水解蛋白酶(酶活力2.4AU/g)、糖化酶(酶活力为1.6×105U/g) 诺维信(中国)生物技术有限责任公司;纤维素酶(酶活力为1.4×106U/g) 湖州米纯生物科技有限公司;硫酸铜、氯化钡、氨水、溴化钾、浓硫酸、无水乙醇、酒石酸钾钠、丙酮、无水乙醚、无水乙二胺等均为分析纯。
1.2 仪器与设备
GB1302电子天平 梅特勒-托利多仪器(上海)有限公司;KDC-1042型离心机 安徽中科中佳科学仪器有限公司;HIS-HA型水浴振荡器 中国哈尔滨市东联电子技术开发有限公司;BPG-9240A型精密鼓风干燥箱、IT-09A-5型恒温磁力搅拌器 上海一恒科学仪器有限公司;PHS-3BW微机型精密酸度计 上海理达仪器厂;DZF-6050型真空干燥箱 上海精宏实验设备有限公司;SX-8-10型箱式电阻炉 天津市泰斯特仪器有限公司;RE 2000A型旋转蒸发仪 上海亚荣生化仪器厂;TU-1901型双光束紫外-可见分光光度计 北京普析通用仪器有限公司;DY-40电动粉末压片机 天津市科器高新技术公司;IR Prestige傅里叶变换红外光谱仪 日本岛津公司。
1.3 方法
1.3.1 原料处理
马铃薯清洗、去皮、切块,加入马铃薯质量50%的纯净水,用组织捣碎机进行机械破碎,破碎粒度60目,将得到的渣浆混合物进行筛分分离,筛上物即为马铃薯渣,将其干燥粉碎、筛分处理,得到可全部通过0.246mm孔径筛的粉末,采用耐高温α-淀粉酶进行酶解脱除残余淀粉,酶解条件为料水比1:15(m/m)、温度90℃、加酶量240U/g(以马铃薯渣干燥样计)、酶解时间30min,酶解后煮沸、维持8min灭酶,冷却至常温,离心沉降,去除上清液,将沉淀物干燥即为马铃薯膳食纤维。
1.3.2 马铃薯膳食纤维基础成分检测
水分含量按照GB 5009.3—2010《食品中水分的测定》规定方法进行;脂肪含量按照GB/T 14772—2008《食品中脂肪的测定》规定方法进行;蛋白质参照GB 5009.5—2010《食品中蛋白质的测定》采用分光光度法进行;灰分参照GB 5009.4—2010《食品中灰分的测定》采用灼烧重量法进行;淀粉参照GB/T 5009.9—2008《食品中淀粉的测定》采用酶水解进行;总膳食纤维、不溶性膳食纤维、可溶性膳食纤维参照GB/T 5009.88—2008《食品中膳食纤维的测定》采用酶质量法进行。
1.3.3 马铃薯膳食纤维成分检测
1.3.3.1 纤维素含量的测定[11]
准确称取0.1g原料于100mL三角瓶中,加入5mL混合酸(硝酸、乙酸体积比为1:10),封口,100℃水浴加热25min;取出冷却至室温,用20mL蒸馏水冲洗三角瓶的瓶壁,与样液一起移入离心管中在3800r/min离心15min,弃去上清液,用适量的蒸馏水冲洗沉淀,3800r/min离心15min,弃去上清液,反复操作3次,将沉淀物转移至250mL具塞碘量瓶中,加入2.5mL浓硫酸和0.1mol/L重铬酸钾溶液10mL,混合均匀,封口,100℃水浴中加热35min,取出冷却后,加入质量分数20%的碘化钾溶液5mL,避光静置5min,取出后加入100mL蒸馏水,用0.21mol/L硫代硫酸钠溶液滴定,待溶液变为黄色时,加入质量分数1%可溶性淀粉1mL,继续滴定,直至溶液变为亮绿色为止,记录消耗硫代硫酸钠溶液的体积,并做空白实验。按照式(1)计算纤维素含量。
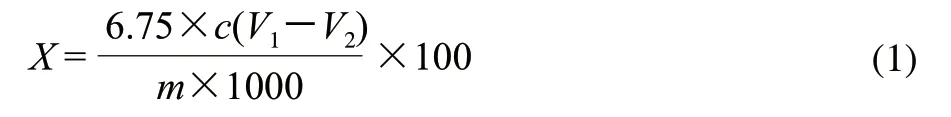
式中:X为纤维素含量/(g/100g);c为硫代硫酸钠的浓度/(mol/L);V1为空白滴定消耗硫代硫酸钠溶液的体积/mL;V2为样液所消耗硫代硫酸钠溶液体积/mL;m为称取原料的质量/g;6.75为纤维素的克当量。
1.3.3.2 半纤维素含量的测定[11]
准确称取0.1g原料于50mL烧杯中,加入质量分数80%的硝酸钙溶液10mL,100℃水浴加热5min,冷却,3800r/min离心分离15min,弃去上清液,用60℃的蒸馏水冲洗沉淀,3800r/min离心分离15min,弃去上清液,反复操作3次;再用适量5mL丙酮冲洗沉淀,静置5min,吸除上清液,反复3次,除去脂类物质。向沉淀中加入2mol/L盐酸10mL,100℃水浴加热45min,使半纤维素水解,冷却后3800r/min离心分离15min,收集上清液。用10mL蒸馏水冲洗沉淀3次,收集上清液,与之前上清液合并于100mL容量瓶中,加水定容至刻度,摇匀后用定性滤纸过滤,收集滤液,用3,5-二硝基水杨酸(DNS)法测定还原糖含量,吸取2mL样液加入DNS试剂1.5mL,100℃水浴加热5min,取出冷却,在波长为540nm处测定其吸光度,对照葡萄糖标准曲线进行分析。同时做空白实验。葡萄糖标准曲线为:y=0.687x+0.017(R2=0.9985)。按照式(2)计算半纤维素含量。
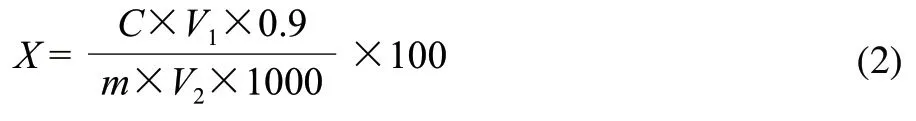
式中:X为半纤维素含量/(g/100g);C为从标准曲线中查得的葡萄糖的含量/mg;V1为提取液定容的体积/mL;V2为比色时吸取样液的体积/mL;m为称取原料的质量/g;0.9为还原糖换算成半纤维素多糖的系数。
1.3.3.3 木质素含量的测定[11]
准确称取0.1g原料于50mL离心管中,加入质量分数1%的醋酸10mL,摇匀后3800r/min离心分离15min,除去上清液,用5mL 质量分数1%的醋酸再将沉淀洗涤一次,同样条件下离心,向沉淀中加入3~4mL乙醇和乙醚混合液(体积比1:1),浸泡沉淀3min,弃去上清液,将沉淀在100℃水浴中蒸干,向沉淀中加入质量分数72%的硫酸3mL,搅拌均匀,室温下静置16h,加入10mL蒸馏水,搅拌均匀,100℃水浴加热5min,冷却后加入5mL蒸馏水和0.5mL质量分数10%的氯化钡溶液,摇匀后3800r/min离心分离15min,沉淀用蒸馏水冲洗2次,然后向沉淀中加入质量分数10%的硫酸和浓度为0.025mol/L重铬酸钾溶液各10mL,100℃水浴加热15min,冷却后,加入质量分数20%的碘化钾溶液5mL,避光静置5min,加入100mL蒸馏水,用0.21mol/L硫代硫酸钠溶液滴定,临近终点时,加入质量分数1%可溶性淀粉1mL,继续滴定,直至滴定终点,记录消耗硫代硫酸钠溶液的体积,并做空白实验。按照式(3)计算木质素含量。
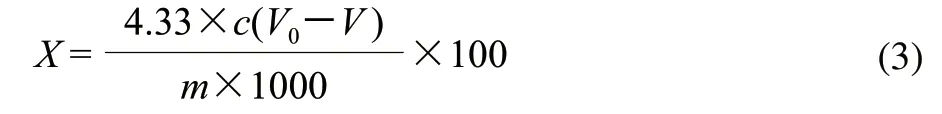
式中:X为木质素含量/(g/100g);c为硫代硫酸钠的浓度/(mol/L);V0为空白滴定消耗硫代硫酸钠溶液的体积/mL;V为样液所消耗硫代硫酸钠溶液体积/mL;m为称取原料的质量/g;4.33为纤维素的克当量。
1.3.3.4 果胶质含量的测定[12]
准确称取5g物料,置于250mL三角瓶中,加入15mL加热至沸的浓度0.05mol/L盐酸,加热回流1h,冷却至室温后定容至250mL,摇匀后用定性滤纸过滤,收集滤液并记录滤液容积数;吸取25mL滤液置于1000mL烧杯中,加蒸馏水至300mL,再加入0.1mol/L氢氧化钠溶液100mL,充分搅拌,放置过夜皂化,脱除甲氧基,生成果胶酸钠。加入1mol/L乙酸溶液50mL,搅拌均匀静置5min,加入浓度0.1mol/L氯化钙溶液25mL,然后边搅拌边滴加2mol/L氯化钙溶液25mL,静置1h后加热5min,趁热过滤,用50℃热水洗涤至不含氯化物为止;用蒸馏水将滤纸上的沉淀冲洗至烧杯中,加热煮沸5min,用G2砂芯漏斗抽滤,于105℃条件下干燥至恒质量。按照式(4)计算果胶质含量。
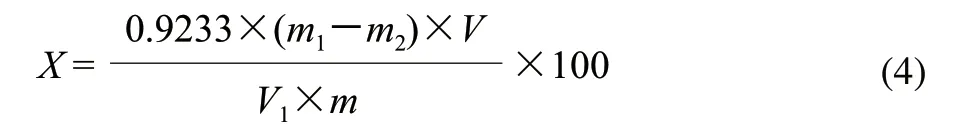
式中:X为果胶酸的含量/(g/100g);V1为消耗提取液的体积/mL;V为提取液总体积/mL;m1为果胶酸钙和砂芯漏斗的总质量/g;m2为砂芯漏斗的质量/g;m为称取原料的质量/g;0.9233为由果胶酸钙换算成果胶酸的系数。
1.3.4 马铃薯膳食纤维物性测定
1.3.4.1 马铃薯膳食纤维膨胀力的测定[13]
准确称取干燥物料0.20g(m),置于5mL量筒中,记录样品在自然堆积情况下的体积(V0),加入蒸馏水至5mL刻度,摇匀后用薄膜密封,室温条件下放置24h,然后测定并记录膳食纤维膨胀体积(V1)。按照式(5)计算马铃薯膳食纤维膨胀力。

1.3.4.2 马铃薯膳食纤维持水力的测定[13]
准确称取1 0 5 ℃条件下干燥至恒质量的物料0.20g(m0),置于50mL烧杯中,向其中加入10mL蒸馏水,封口后室温条件下浸泡12h,然后用滤纸滤除水分,称量湿样的质量(m1)。按照式(6)计算马铃薯膳食纤维持水力。

1.3.4.3 马铃薯膳食纤维的持油力[13]
准确称取干物料0.20g(m0),放入50mL离心管中,加入豆油10mL,使之混合均匀,密封后37℃条件下静置1h,3900r/min条件下离心15min,弃去上层油脂,称量剩余残渣的湿质量(m1)。按照式(7)计算马铃薯膳食纤维持油力。
1.3.5 马铃薯膳食纤维平均相对分子质量的测定
1.3.5.1 马铃薯膳食纤维-铜乙二胺溶液的制备[14]
称取2g马铃薯膳食纤维,置于105℃烘干箱中干燥30min,取出后放在干燥器中冷却20min。准确称取0.040g干燥的马铃薯膳食纤维,置于100mL烧杯中,加入适量铜粒(铜粒在使用前依次用浓硝酸、无水乙二胺溶液浸泡1h,再用蒸馏水反复洗涤至中性)、再加入10mL铜乙二胺溶液、10mL蒸馏水,用300r/min磁力搅拌器搅拌60min,使其完全溶解,配成质量浓度为0.2g/100mL的马铃薯膳食纤维-铜乙二胺溶液。
1.3.5.2 马铃薯膳食纤维聚合度的测定及相对分子质量计算[14]
准确吸取10mL马铃薯膳食纤维-铜乙二胺溶液于经润洗处理的乌式黏度计中,置于恒温水浴锅中,垂直固定,25℃恒温测定黏度。取3次平均值为最终结果,两次测量值的差应小于较小值的0.4%。
对于非牛顿流体,黏滞阻力的变化影响黏滞阻力与速度梯度的比值,高聚物在稀溶液中的黏度值反映了液体流动时内摩擦力的大小,将溶液无限稀释可减弱或消除高聚物分子间的内摩擦。当液体在毛细管黏度计中因重力作用而流出时,相对黏度(ηr)、特性黏度([η]’)、聚合度(DP)之间关系可遵循文献[15],即式(8)、(9)、(10)。按式(8)计算相对黏度(ηr)。

式中:t、t0为聚合物溶液及溶剂在毛细管内的流出时间/s;K为黏度计的校正系数。
由式(8)可得出ηr,从文献[15]中可查出ηr所对应的[η]×c值,根据式(9),可计算出[η]’。

式中:[η]’为高聚物溶液特性黏度;c’为马铃薯膳食纤维-铜乙二胺溶液的实际质量浓度/(g/100mL)。
高聚物平均聚合度Dp与[η]’之间的关系可用式(10)表示:

平均分子质量M与聚合度之间的关系为:

1.3.6 马铃薯膳食纤维红外光谱
用纤维素酶对马铃薯膳食纤维进行酶解,酶解条件为:料水比1:10(m/m)、温度45℃、加酶量7000U/g(以马铃薯渣干燥样计)、调节pH4.5,酶解时间120min,酶解后煮沸、维持8min灭酶,冷却至常温,离心沉降,沉淀烘干粉碎得到马铃薯不溶性膳食纤维;上清液经浓缩后加入其4倍体积的体积分数为95%乙醇溶液进行醇沉,在3500r/min的条件下离心10min,离心后将沉淀干燥粉碎,即得马铃薯可溶性膳食纤维。称取干燥后的马铃薯可溶性与不溶性膳食纤维各2mg、分别与200mg溴化钾粉末混合均匀,然后在压力8MPa条件下进行压片。扫描范围为400~4000cm-1。
2 结果与分析
2.1 马铃薯膳食纤维的基础成分

表1 马铃薯膳食纤维基础成分含量Table 1 Basic chemical components of potato dietary fiber
由表1可知,马铃薯膳食纤维中含有多种成分,其中总膳食纤维含量在80g/100g以上,可溶性膳食纤维含量为4.98g/100g。有研究报导木瓜渣的基本成分中总膳食纤维的含量为69.58g/100g可溶性膳食纤维含量6.97g/100g[16];甘薯渣中总膳食纤维27.40g/100g,可溶性膳食纤维含量2.66g/100g[17];大豆皮总膳食纤维含量为73.31g/100g[18];玉米皮总膳食纤维含量为60.00g/100g左右[19],玉米皮中可溶性膳食纤维含量为3.97g/100g[13],大豆皮中可溶性膳食纤维的含量为0.79g/100g[18]。由此可以看出马铃薯膳食纤维中总膳食纤维和可溶性膳食纤维的含量均较高,是一种较好的膳食纤维资源。
2.2 马铃薯膳食纤维的组分

表2 马铃薯膳食纤维组分含量Table 2 Firber components of potato dietary fiber
由表2可知,马铃薯膳食纤维中纤维素和半纤维素的含量均较高。半纤维素及果胶质含量均高于文献报道的甘薯膳食纤维及大豆皮膳食纤维,而木质素含量低于甘薯及大豆膳食纤维[17]。因此马铃薯膳食纤维具有更好的柔性及较低的相对分子质量,将是生产高品质膳食纤维的良好原料。纤维素及半纤维素具有预防便秘、调节血糖、降低胆固醇的作用;果胶质可赋予被加工物料良好的胶凝和乳化稳定性能,还具有抗菌、消肿、解毒、降血脂、抗辐射等作用[20]。因而马铃薯膳食纤维更多的调节人体异常代谢功能还需进一步研究。
2.3 马铃薯膳食纤维的物性
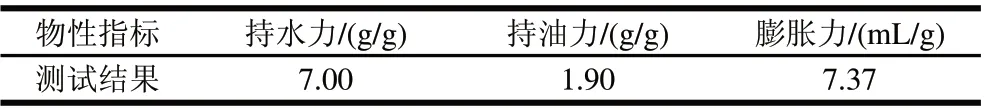
表3 马铃薯膳食纤维物性Table 3 Physical properties of potato dietary fiber
由表3可知,马铃薯膳食纤维具有较好的持水力和膨胀力,均高于玉米皮纤维、大豆皮纤维及脱脂米糠[21]。膨胀力高于甘薯膳食纤维,但持水力及持油力略低于甘薯膳食纤维[17]。马铃薯膳食纤维较好的持水力和膨胀力有利于其在抗便秘、改善肠道环境及预防肥胖等方面发挥作用[22]。
2.4 马铃薯膳食纤维的聚合度和平均相对分子质量

表4 聚合度和平均相对分子质量测定结果Table 4 Polymerization degree and average molecular weight of potato dietary fiber
由表4可知,马铃薯膳食纤维聚合度和平均相对分子质量较小,与其具有较高含量的可溶性膳食纤维及较强的吸水膨胀能力相符。有研究表明:豆渣水溶性膳食纤维的相对分子质量高达546673[23],一般情况下,相对分子质量越小,分子聚合度越低,物质的溶解性越好[23],越容易功能化处理,因此马铃薯膳食纤维的功能化处理难度将低于玉米皮纤维及大豆皮纤维,其在高纤维食品生产中的应用前景也更加广阔。
2.5 马铃薯膳食纤维的结构特性
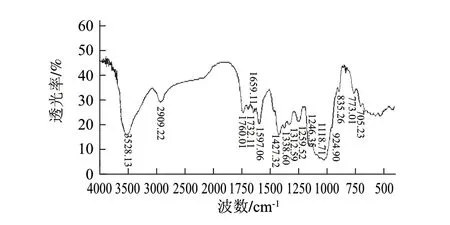
图 1 马铃薯可溶性膳食纤维的红外图谱Fig.1 Infrared spectrum of soluble potato dietary fi ber
由图1可知,马铃薯可溶性膳食纤维具有多糖的特征吸收峰。3528cm-1处为O-H的伸缩振动,2909cm-1处为糖类亚甲基上C-H的伸缩振动,1338、1259、1246cm-1处为C-H的变角振动,这些吸收峰都为糖类的特征吸收峰。1427cm-1处的吸收峰为O-H的变形振动引起的;1118cm-1和1053cm-1处的吸收峰是由两种C-O伸缩振动所引起的,一种是属于C-O-H的,另一种是糖环的C-O-C[24];1312cm-1处为羧酸的C-O伸缩振动,在924cm-1处为成键的O-H面外弯曲振动,证明马铃薯可溶性膳食纤维中存在羧酸二聚体;1659、1732、1760cm-1处的吸收峰是羰基的吸收峰,说明马铃薯可溶性膳食纤维中存在糖醛酸[25];1597cm-1处的吸收峰是由C=C骨架伸缩振动引起的;835cm-1处出现弱小尖峰,表明马铃薯可溶性膳食纤维的中存在α-型吡喃糖[26];在773cm-1处有吸收峰,说明组成的结构中含有α-D-木吡喃糖[27];705cm-1处的吸收峰是由C-H的变形振动所引起的。
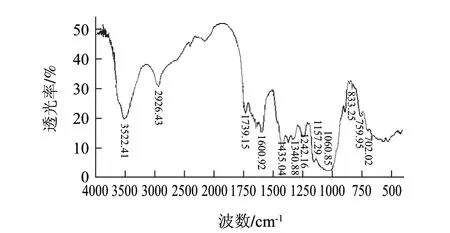
图 2 马铃薯不溶性膳食纤维的红外图谱Fig.2 Infrared spectrum of insoluble potato dietary fi ber
马铃薯不溶性膳食纤维包括半纤维素、纤维素以及木质素。由图2可知,马铃薯不溶性膳食纤维在3522cm-1处有一个较宽且吸收较强的吸收峰,这是羟基的特征峰,说明马铃薯不溶性膳食纤维中存在着处于缔合状态的氢键[24]。2926cm-1处为糖类亚甲基上C-H的伸缩振动,1340、1242cm-1处为C-H的变角振动,这些吸收峰都是糖类的特征吸收峰。1739cm-1处为半纤维素C=O的伸缩振动,1157cm-1是半纤维素和纤维素C-O-C伸缩振动,1060cm-1是半纤维素和纤维素中C-O的伸缩振动,这与纤维素的结构相符。1600cm-1处的吸收峰为C=C骨架伸缩振动引起的;1435cm-1处的小尖锋是O-H的变形振动;833cm-1处出现了吸收峰表明马铃薯不溶性膳食纤维的单糖中含有α-型吡喃糖[26];759cm-1处为环外的C-H弯曲振动,说明组成中存在α-D-木吡喃糖[27];702cm-1处的吸收峰是由C-H的变形振动引起的。
3 结 论
3.1 马铃薯膳食纤维的平均相对分子质量为170333,聚合度为1051;总膳食纤维含量高达81.50%,其中可溶性膳食纤维含量为4.98%,持油力为1.9g/g,持水力为7.00g/g,膨胀力为7.37mL/g。综合品质优于未经功能化处理的玉米皮纤维及大豆皮纤维。
3.2 马铃薯膳食纤维的红外光谱图显示其具有典型的糖类特征吸收峰C=O键、C-H键、COOR和游离的·O-H,单糖组分结构中有吡喃环结构;存在糖醛酸和羧酸二聚体。
[1] 李建文, 杨月欣. 膳食纤维定义及分析方法研究进展[J]. 食品科学, 2007, 28(2): 350-355.
[2] 温志英, 杨丽钦. 花生壳水溶性膳食纤维微波辅助提取工艺及其性质研究[J]. 中国粮油学报, 2011, 26(4): 99-103.
[3] ZHANG Min, BAI Xin, ZHANG Zesheng. Extrusion process improves the functionality of soluble dietary fiber in oat bran[J]. Journal of Cereal Science, 2011, 54: 98-103.
[4] ZHANG N, HUANG C H, OU S Y. in vitro binding capacities of three dietary fibers and their mixture for four toxic elements, cholesterol, and bile acid[J]. Journal of Hazardous Materials, 2011, 186(1): 236-237.
[5] GALISTEO M, DUARTE J, ZARZUELO A. Effects of dietary fibers on disturbances clustered in the metabolic syndrome[J]. The Journal of Nutritional Biochemistry, 2008, 19(2): 71-84.
[6] ESPOSITO F, ARLOTTI G, BONIFATI A M, et al. Antioxidant activity and dietary fibre in durum wheat bran by-products[J]. Food Research International, 2005, 38(10): 1167-1173.
[7] 陈雪峰, 张振华, 王锐平. 苹果膳食纤维制备中水溶性膳食纤维变化的研究[J]. 食品科技, 2010, 31(8): 117-120.
[8] 潘利华, 徐学玲, 罗建平. 超声辅助提取水不溶性大豆膳食纤维及其物理特性[J]. 农业工程学报, 2011, 27(9): 387-392.
[9] 杨希娟. 马铃薯渣开发利用前景分析[J]. 粮食加工, 2009, 34(6): 68-70.
[10] 吕金顺, 韦长梅, 徐继明, 等. 马铃薯膳食纤维的结构特征分析[J]. 分析化学简报, 2007, 35(3): 443-446.
[11] 王金主, 王元秀, 李峰, 等. 玉米秸秆中纤维素、半纤维素和木质素的测定[J]. 山东食品发酵, 2010(3): 44-47.
[12] 陈毓荃. 生物化学实验法和技术[M]. 北京: 科学出版社, 2002: 180-183.
[13] 王大为, 郭雪飞, 杨羿. 高温高压挤出处理对玉米皮膳食纤维溶解特性及物性的影响[J]. 食品科学, 2011, 32(13): 84-88.
[14] HG/T 2758—1996 乙酸纤维素稀溶液黏数和黏度比的测定[S]. 北京:中华人民共和国化学工业部, 1996.
[15] GB 5888-1986 苎麻纤维素聚合度测定方法[S]. 北京: 国家标准局, 1986.
[16] 罗欢. 木瓜渣膳食纤维的分离提取及理化性质研究[D]. 重庆: 西南大学, 2010.
[17] 曹媛媛. 甘薯膳食纤维的制备及其物化特性的研究[D]. 乌鲁木齐: 新疆农业大学, 2007.
[18] 肖安红. 常见膳食纤维微粉的研究[D]. 武汉: 华中科技大学, 2008.
[19] 王言. 玉米皮膳食纤维的制备、功能活化及活性的研究[D]. 长春: 吉林大学, 2010.
[20] 万建华. 马铃薯果胶的提取及其性质的研究[D]. 无锡: 江南大学, 2008.
[21] 张艳荣, 丰艳, 孙丽琴, 等. 响应面法优化米糠挤出工艺及其物性研究[J]. 食品科学, 2010, 31(20): 146-151.
[22] 戚勃, 李来好. 膳食纤维的功能特性及在食品工业中的应用现状[J]. 现代食品科技, 2006, 22(3): 272-279.
[23] 钟艳萍. 水溶性膳食纤维的制备及其性能的研究[D]. 广州: 华南理工大学, 2011.
[24] 刘春兰. 少数民族地区药用植物多糖的化学与药理[M]. 北京: 中央民族大学出版社, 2008: 39-41.
[25] 刘志广, 张华, 李亚明. 仪器分析[M]. 大连: 大连理工大学出版社, 2007: 273-292.
[26] SHI Ying, SHENG Jianchun, YANG Fangmei, et al. Purification and identification of polysaccharide derived from Chlorella pyrenoidosa[J]. Food Chemistry, 2007, 103(1): 101-105.
[27] 郭振楚. 糖类化学[M]. 北京: 化学工业出版社, 2005: 106-119.
