Quantum Chemistry of PAHs Thermal Cracking with Different Hydrogenation Degree
2013-07-25WangChunluZhouHanDaiZhenyuZhaoXiaoguangZhaoYi
Wang Chunlu; Zhou Han; Dai Zhenyu; Zhao Xiaoguang; Zhao Yi
(Research Institute of Petroleum Processing, SINOPEC, Beijing 100083)
Quantum Chemistry of PAHs Thermal Cracking with Different Hydrogenation Degree
Wang Chunlu; Zhou Han; Dai Zhenyu; Zhao Xiaoguang; Zhao Yi
(Research Institute of Petroleum Processing, SINOPEC, Beijing 100083)
In order to investigate the influence of hydrogenation degree and structural variety on reaction trend of polyaromatic hydrocarbons (PAHs) in resins and asphaltenes portion of heavy oil, a series of PAHs with different hydrogenation degree were selected as model compounds to simulate their different hydrogenation stage, and the PAHs thermal cracking reaction was simulated based on free radical mechanism by the density functional theory (DFT) to search for reactions’ transition state. By comparing the dynamic data obtained from reaction simulation, it is showed that processing difficulty could rise with increasing condensed aromatic ring number, and hydrogenation could promote ring cleavage reaction, but excessive hydrogenation would decrease the oil conversion rate to reduce light-end products. In conclusion, proper hydrogenation was quite critical in promoting light-end products conversion efficiency and saving the processing cost as well. Operational instructions were given based on both PAHs hydrogenation performance and conclusions were drawn up from reaction simulation results.
heavy oil; PAHs; thermal cracking; ring cleavage; molecular modeling; DFT
1 Introduction
Nowadays crude oil is becoming heavier worldwide, so there is an urgent need to optimize heavy oil processing technology in order to increase the yield of lightend products and to remove heteroatoms in finished oil products, which is of great importance to meet both the needs of economic requirements and environmental regulations. To solve this problem, the average molecular weight of heavy oil must be decreased efficiently. Hydrotreating technology can make oil feedstock react on hydrogen to remove contaminants such as sulfur and nitrogen, and at the same time saturate aromatics and olefins to a certain extent[1]. Besides those advantages of hydrotreating technology listed above, the processing cost is relatively high compared with decarbonizing technology. How to balance this contradiction between the needs to maximize light ends conversion and the economic requirements for low-cost technology for manufacturing light liquid product has become a major goal of today’s oil refining research. In other words, it is necessary to know how to prevent coking while achieving high utilization of hydrogen resource during hydrotreating process.
Polyaromatic hydrocarbons (PAHs) that are present in resins and asphaltenes portion of heavy oil are known as the main precursor of coke[2-3], also, they are considered to be not easily converted during thermal cracking and hydrogenation reactions[4]. Thus PAHs with different aromatic rings were selected as model compounds and the DMol3module of Material Studio was adopted to simulate thermal cracking reaction of PAHs in order to identify the influence of PAHs’ structure on reaction trend. Radical reaction mechanism was applied in this paper for thermal cracking reaction. Then the molecular modeling calculation method was applied to reactions to investigate their reaction trends, and by studying the hydrogenation performance described in a previous paper[5]and comparing the obtained dynamic data, the instructions for operation optimization during hydrotreating process were mapped out.
2 Computational Methods
2.1 Selection of model compounds
PAHs selected as model compounds were composed of two to seven aromatic rings as shown in Table 1. These structures can be categorized into cata-condensed and peri-condensed PAHs and are listed into two rows. As shown in the left row, the cata-condensed PAHs are molecules that have only two atoms and one bond in common. On the other hand, PAHs with ring cata-condensed to different sides of two other rings that are themselves catacondensed together are said to possess the peri-condensed structure[6].

Table 1 PAHs model compounds consisting of 2 to 7 aromatic rings
2.2 Modeling method and reaction design
DMol3is one module of the Material Studio software which uses DFT to simulate chemical reaction rapidly and accurately. Geometry and transition state optimizations are performed using delocalized internal coordinates, suited for both molecular[7]and periodic[8]calculations. DMol3achieves its speed and accuracy by using numerical functions on an atom-centered grid as its atomic basis[9-10].
This study is proposed by DMol3module to simulate the thermal cracking of model PAHs in order to identify their reaction trends. The GGA-BLYP function was used, with the DNP basis set properly chosen. Both the structures of reactant and product were first optimized with DMol3module within the DFT framework. Then the transition state search was carried out by TS Search task and verified with vibrational analysis. The transition state of thermal cracking reaction was verified by animating the only imaginary normal mode associated with the reaction coordinate.
The example of designed thermal cracking reaction is shown in Figure 1.
3 Results and Discussion
3.1 Comparison of thermal cracking reactions
Reaction simulation data are shown in Table 2.
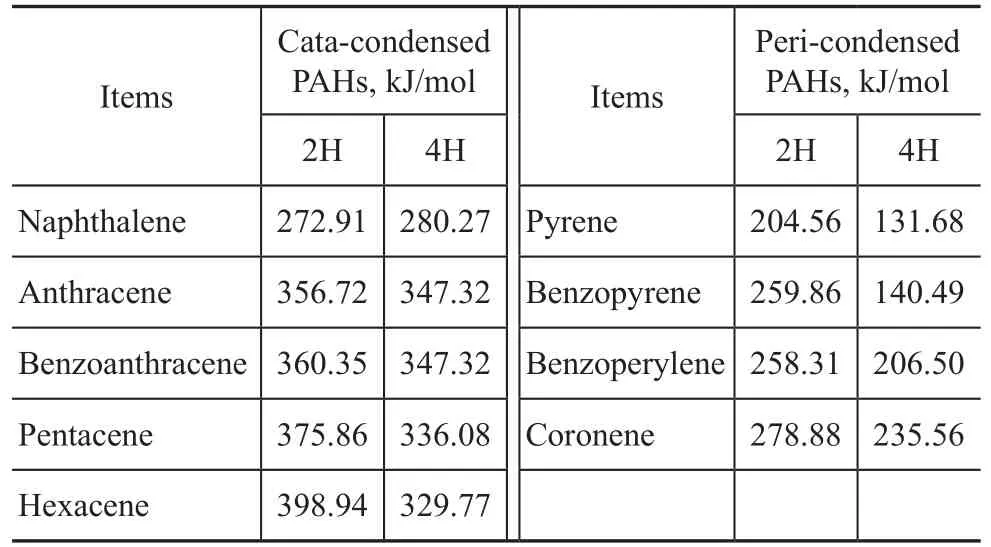
Table 2 Barrier of energy scenario on thermal cracking simulation data of model compounds
The reaction trends of PAHs thermal cracking at differenthydrogenated levels were sketched based on simulation data as shown in Figure 2.

Figure 1 Thermal cracking designed for 1,2,3,4-tetrahydro-anthracene
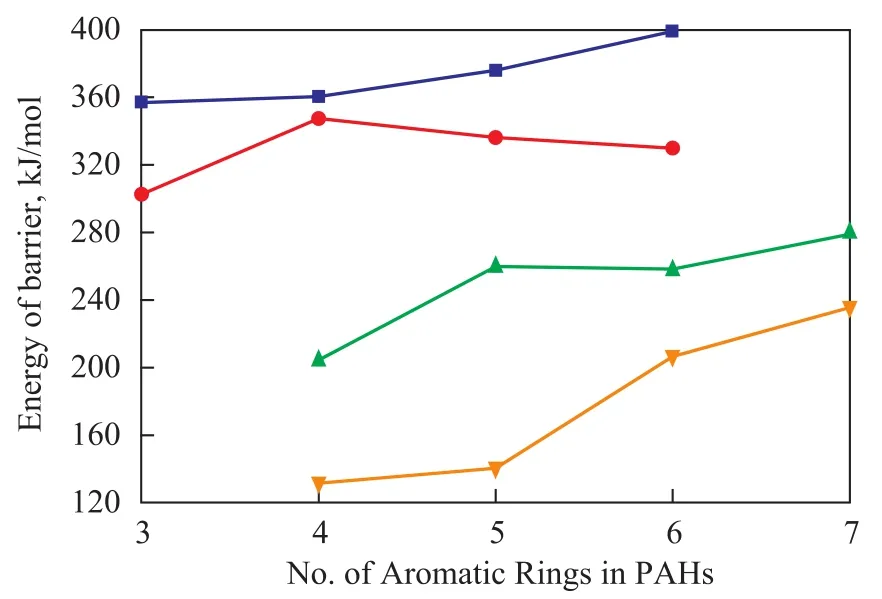
Figure 2 Energy of barrier comparison of PAHs thermal cracking
3.2 Discussion of results
It can be seen from Figure 2 that the energy of barrier rises with an increasing number of aromatic rings that are present in PAHs structure, which means that PAHs thermal cracking becomes more difficult when condensed aromatic ring number increases. This is because the aromatic ring is a kind of rigid group which would contribute to the rigidity of the whole molecular structure. As a result, PAHs’ rigidity increases with an increasing number of aromatic rings comprised in its structure. The energy required for molecular torsional deformation when ring cleavage takes place is affected by structure rigidity. When structural rigidity increases, barrier energy rises as well. Thus through comparison of barrier energy, which is the minimum energy required for a chemical reaction, could reveal the processing difficulty of heavy oil. The heavier the oil feedstock is, the more difficult the oil processing would be.
With respect to the cata-condensed PAHs, their barrier energy for thermal cracking reaction is much higher than the peri-condensed PAHs with the same number of aromatic rings, which can also be explained by molecular rigidity theory. The deformation ability of peri-condensed PAHs to acquire such dish-like structure is relatively lower than cata-condensed PAHs, which means that when ring cleavage occurs, the peri-condensed PAHs would need a relatively lower energy for torsional deformation before molecule’s hyperconjugation structure is completely destructed. This can be observed from the optimized thermal cracking products of coronene as shown in Figure 3, and both products retain the flat-dishlike structure after certain level of hydrogenation progress, as compared with products of 1,2,3,4-tetrahydro-anthracene that are shown in Figure 1.

Figure 3 Ring cleavage products of coronene hydrogenated with 2 H and 4 H atoms
The third conclusion drawn from this research is that the influence of hydrogenation degree on thermal cracking of the cata-condensed PAHs is relatively smaller than the peri-condensed PAHs. This phenomenon is ascribed to their different hydrogenation progress and structure character.
During hydrogenation of cata-condensed PAHs, hydrogen atoms first attack at the middle aromatic ring of PAHs molecule, followed by switching of subsequent hydrogenation locations to the outside rings of PAHs structure[5]. Since the location that ring cleavage reaction tends to occur at the bond which has been saturated with hydrogen,in the case of cata-condensed PAHs that are hydrogenated with 2 hydrogen atoms, thermal cracking can only possibly lead to cleavage of molecules at the middle ring, thus affecting the hyperconjugation structure of the two adjacent aromatic rings, whereas in the case of catacondensed PAHs that have an even higher hydrogenation degree, ring cleavage would be switched to side chains and therefore the hyperconjugation structure that has been affected is limited at the end of molecule.
The hydrogenation process of peri-condensed PAHs is different from that of cata-condensed PAHs. Hydrogen is first routed to saturate the outer carbons of PAH molecule, which would cause its hyperconjugation structure much more sensitive to hydrogenation reaction. Consequently, when hyperconjugation structure is destructed, energy of barrier for thermal cracking reaction is greatly reduced.
4 Conclusions
Based on simulations of model compounds’ thermal cracking reactions, several conclusions can be drawn up from above-mentioned results:
(1) The difficulty of thermal cracking reaction rises with the increasing number of aromatic rings that are present in PAHs structure, indicating that the heavier the crude oil is, the more difficult the oil processing would be. In this case, an increase in processing temperature might be needed.
(2) The approach of thermal cracking is to increase the yield of liquid products and thus to reduce the average molecular weight of heavy oil, which is also closely related with the bond location where ring cleavage takes place. Based on the nature of cata-condensed PAHs hydrogenation process, it is possible to utilize certain hydrogenation stage to promote the efficiency of light ends conversion. Also, the hydrogenation simulation data show that hydrogenation trend is not affected by neither molecule weight nor hydrogenation degree, and the general energy of barrier of hydrogenation is quite low which could be considered to be associated with an elementary reaction, and eventually it is possible to inhibit PAHs condensation reaction provided that PAHs can keep in contact with radical hydrogen effectively.
(3) By referring to practical process operation, the increase in hydrogen partial pressure and introduction of a real-time analysis system into reaction system at the initial hydrotreating stage might be needed in order to control the level of hydrogenation degree. When certain level of hydrogenation is reached, hydrogen partial pressure could be reduced, and likewise, a proper feedstock hydrogenation extent could cut down the oil processing cost.
Acknowledegement:This work was sapported by China Petrochemical Corporation (SINOPEC) (Contact No.112101).
Reference
[1] Girgis M J, Gates B C. Reactivities, reaction networks, and kinetics in high-pressure catalytic hydroprocessing[J]. Ind Eng Chem Res, 1991, 30(9): 2021-2058
[2] Chen Guimei, Zhang Xiangwen, Mi Zhentao. Effects of pressure on coke and formation of its precursors during catalytic cracking of toluene over USY catalyst[J]. Fuel Chemistry and Technology, 2007, 35(2): 211-216
[3] Alvarez P, Díez N, Santamaría R, et al. Novel coal-based precursors for cokes with highly oriented microstructures[J]. Fuel, 2012, 95: 400-406
[4] van Rheinberg O, Lucka K, Köhne H. About the process improvement of adsorptive desulphurisation by adding hydrogen donators as additives in liquid fuels[J]. Journal of Power Source, 2011, 196(21): 8983-8993
[5] Wang Chunlu, Zhou Han, Wang Zijun, et al. Molecular modeling of direct radical hydrogenation on different PAHs molecules [J]. Computer and Applied Chemistry, 2012 29(10): 1221-1224 (in Chinese)
[6] Moss G P. Nomenclature of fused and bridged fused ring system [J]. Pure & Appl Chem, 1998, 70(1): 143-216
[7] Baker J, Kessi A, Delley B. The generation and use of delocalized internal coordinates in geometry optimization [J]. J Chem Phys, 1996, 105 (1): 192-212
[8] Andzelm J, King-Smith R D, Fitzgerald G. Geometry optimization of solids using delocalized internal coordinates [J]. Chem Phys Lett, 2001, 355 (3/4): 321-326
[9] Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules[J]. J Chem Phys, 1990, 92(1): 508-517
[10] Delley B. Fast calculation of electrostatics in crystals and large molecules[J]. J Phys Chem, 1996, 100 (15): 6107-6110
Recieved date: 2013-01-28; Accepted date: 2013-04-18.
Zhao Yi, Telephone: +86-10-82368079; E-mail: zhaoyi.ripp@sinopec.com.
杂志排行

中国炼油与石油化工的其它文章
- Preparation and Catalytic Performance of Potassium Titanate Used as Soot Oxidation Catalyst
- A Probe into Process for Maximization of Low-carbon Olefins via Co-processing of Methanol and Heavy Oil
- Influence of Carbon Content on S Zorb Sorbent Activity
- Propylene Polymerization Catalysts with Sulfonyl Amines as Internal Electron Donors
- Isolation and Characterization of a Thermophilic Oil-Degrading Bacterial Consortium
- Research on Catalytic Properties of Palladium Catalyst Prepared by Biological Reduction Method