氧化对脂质体膜性质的影响
2013-07-17耿亚男聂新艳杨贝贝
耿亚男,曹 栋,聂新艳,杨贝贝
(江南大学食品学院,江苏无锡 214122)
脂质体是一类由脂质发生自我凝聚时产生的微囊体。近些年来,脂质体在食品、化妆品、医药中都有广泛应用[1],如抗癌药物的靶向给药,包埋激素类肽、抗生素、维生素、酶等,脂质体已经成为一种理想的药物传递载体。脂质体的主要膜材为磷脂,磷脂是一种常见的有机两性分子,由于其脂肪酸不饱和程度较高,极易发生氧化。在实际生产过程中,脂质体的放大性生产及其保存是阻碍其应用的最大难题。影响脂质体保存的主要原因是其具有易氧化、变形、分解等不稳定的缺点,其中,磷脂的氧化是影响脂质体品质的最大因素。氧化后的磷脂必然导致膜结构发生改变,会影响双层膜的流动性,使其发生相分离[2-3],并使囊泡内部无序化,形成孔洞[4-5],从而破坏脂质体。究其氧化原因,主要是由于活性氧攻击脂质体膜,氧化后的磷脂双键被破坏而形成各种不同的小分子[6]。目前,磷脂氧化对脂质体膜的影响机制还不明朗,本实验通过Fenton反应控制脂质体氧化,并通过脂质体的粒径、Zeta电位、脂质体膜的微极性、流动性及形态等,研究了氧化对脂质体膜的破坏作用。
1 材料与方法
1.1 材料与仪器
大豆磷脂酰胆碱 SPC90,江苏曼氏生物科技有限公司;1,6-二苯基-1,3,5-己三烯 DPH Sigma公司;钙黄绿素 上海迈坤化工有限公司;Sephadex G-25 北京瑞达恒辉科技发展公司;氯仿、甲醇、氯化亚铁、过氧化氢、磷酸二氢钠、磷酸氢二钠、硫代巴比妥酸、三氯乙酸、浓盐酸 国药集团化学试剂有限公司;所用试剂均为分析纯。
UV-260紫外分光光度计 日本岛津;纳米粒度及ZETA电位仪 英国马尔文公司;F-7000荧光光谱仪;H-7650透射电子显微镜 日本Hitachi公司;Hettich EBA20小型离心机 莱比信科技发展有限公司;电子精密天平 上海梅特勒-托利多仪器有限公司;旋转蒸发仪 南京予凯仪器设备有限公司;超声波发生器 无锡科洁超声电子设备有限公司。
1.2 实验方法
1.2.1 脂质体制备 取0.02g大豆卵磷脂置于小烧杯中,加入3mL氯仿/甲醇(2/1,V/V)混合液,放入1.0g左右玻璃珠,晃动溶解。将混合液转移到500mL烧瓶中,于45℃、40r/min水浴蒸发氯仿/甲醇混合有机溶剂。在瓶内壁形成一层薄膜,持续真空旋蒸1h以除去残余溶剂,结束后冲入保护气体氮气。加入5mL磷酸盐缓冲液(10mmol/L,pH7.4)水化脂质薄膜,形成5mL粗脂质乳状液。暗处用磁力搅拌器搅拌10min,所得粗脂质体超声(200W)处理10min,使用0.22μm滤膜过滤两次整粒,暗处放置1h进行下步操作。
1.2.2 MDA值的测定 磷脂氧化后产物MDA的测量根据文献[7]并稍有修改。TBA溶液配制:TCA 30g,TBA 0.75g,0.25mol/L 盐酸 200mL,温热溶解,过滤后得TCA-TBA-HCl溶液。量取脂质体乳状液1.0mL于10mL带刻度试管中,加入TCA-TBA-HCl混合溶液5mL,混匀,于100℃水浴30min,立刻冷却。使用 TCA-TBA-HCl溶液定容至10mL,5000r/min离心 10min,以 TCA-TBA-HCl溶液为空白,测定532nm处溶液的吸光值(MDA的摩尔吸光率为1.56×105L/mol·cm)。刚制备好的脂质体即用TBA的方法测量其在制备过程中的氧化程度。
1.2.3 透射电镜观察脂质体形态 参照Mohsen M等[8]磷钨酸-负染色法预处理脂质体乳状液,将体系中磷脂酰胆碱的浓度稀释至为10mg/mL,先用洁净干燥的注射器吸取刚制备好的脂质体滴至附有支持膜的铜网上,样品在Formvar膜上吸附约10min后,用滤纸小心吸除多余液体,再向膜上滴加2%磷钨酸,染色5min左右,滤纸吸去染液,用灯烘干。在Hitachi透射电镜(H-7650)下观测并调整适合放大倍数进行拍照。
1.2.4 脂质体膜的微极性测定 制备10-4mol/mL的芘的甲醇溶液,取0.5mL芘溶液于三角瓶中,氮气吹干,加入40mL所制备的粗脂质体乳状液,控制温度40℃超声(200W)1h,测量芘的第一发射峰与第三发射峰强度比值I1/I3(在372nm处荧光强度与在384nm处的比值)以表征脂质体膜的微极性。
1.2.5 脂质体包封率测定 将适量钙黄绿素溶于磷酸盐缓冲液中,用上述缓冲液制备脂质体。取1mL脂质体样品,置于用PBS平衡过的sephadex G-25柱上端,以PBS为洗脱液,于206nm处监测脂质体流出时间,使用荧光分光光度计测量荧光强度(EM 490nm,EX 520nm),收集脂质体。将流出的脂质体溶液置于25mL容量瓶中,PBS加至刻度,混匀。测定分离前后脂质体的荧光强度(F),分别计算两种脂质体的包封率,按公式(1)计算。

式中,E(%)包封率;K-样品的稀释倍数比,本实验为K取25;FL-所分离脂质体的荧光值;FT-原脂质体总荧光强度[9]。
1.2.6 脂质体的膜流动性测定 使用1,6-二苯基-1,3,5-己三烯(DPH)对囊泡双层流动性进行评估,测定双层膜的荧光偏振率。DPH储备液添加至脂质体溶液中,达到最终磷脂与DPH浓度(molar)比为500∶1。混合液进行超声(200W)并且45℃水浴保持1h。使用荧光光谱仪(F-7000)测量荧光光谱,设定发射光波长为426nm,激发光波长为348nm,发射光狭缝宽度10nm,激发光狭缝宽度为5nm。荧光偏振率按式(2)、式(3)计算。


式中,对公式中的字母含义做解释;计算出相对偏振度Pn/P0[10],P0为所测偏振度几个之中的最小值(即氧化24h时所测值),Pn为除最小值之外的其他值。计算的偏振率越大,膜的流动性越小,两者成反比。
2 结果与讨论
2.1 脂质体MDA值的变化
图1为未添加Fenton试剂和添加Fenton试剂后24h内脂质体MDA值的变化。
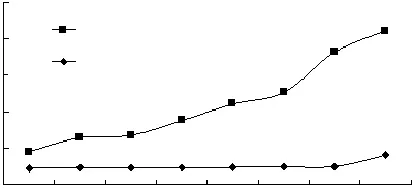
图1 Fenton试剂对脂质体氧化的影响Fig.1 Influence of oxidation on MDA of liposomes
分析图1可知,与未添加Fenton试剂的脂质体相比,添加Fenton试剂后脂质体的氧化程度明显增加,并且在24h内,氧化已经达到一定程度。因此,可以确定添加Fenton试剂能够迅速引发脂质体氧化,并且通过重复实验可知,脂质体的氧化程度可以通过控制其反应时间来控制。
2.2 脂质体粒径及Zeta电位的变化
脂质体溶液适度稀释后测量其粒径。用纳米粒度及Zeta电位仪测其粒径分布,结果见表1和图2。
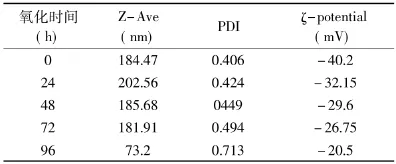
表1 脂质体氧化后粒径及Zeta电位的变化Table1 The influence of oxidation on particle size and Zeta potential
由图2可以看出,在未加入Fenton试剂时,脂质体粒径分布为正态分布,且分布比较集中。添加Fenton试剂的脂质体与未添加Fenton试剂的脂质体相比,在24h后,脂质体的粒径略微变大,PDI指数也有所增加,这可能是脂质体氧化后膜之间发生融合而造成的。未添加Fenton试剂的脂质体粒径与其24h前相比,粒径稍有减小,这是刚制备好的脂质体在抽滤整粒后多聚集在一起,随后放置过程中发生少量分离的原因。表1结果显示,脂质体在氧化过程中,其粒径变化趋势为先增大再变小。氧化72h内,脂质体大小并发生较明显变化,但96h时,脂质体的粒径及分布均发生剧烈改变,此时的脂质体膜可能由于氧化发生崩解,脂质体变成膜的碎片,导致平均粒径变小,且大小不一。

图2 脂质体氧化后粒径的变化Fig.2 The changes of liposome size after oxidation
脂质体膜表面电荷对脂质体的物理化学稳定性也具有重要意义[11],如改善脂质体降解,及降低磷脂氧化率。添加Fenton试剂后用纳米粒度及Zeta电位仪测脂质体膜表面电位,由表1可以看出,Fenton试剂加入后,Zeta电位的绝对值减小。一般来说,脂质体膜电位绝对值越大,由于膜之间的斥力作用,体系越加稳定。由文献[12]可知,实验原料中还有少量电负性磷脂,因此,Zeta电位的降低一方面可能是由于电负性磷脂氧化导致含量降低,另一方面可能是氧化所产生的氧化产物而造成的。另外,Zeta电位绝对值的降低使得体系更加不稳定。
2.3 电镜扫描脂质体形态的变化
由图3分析可知,所制备的脂质体大小较为均匀,大小基本保持在110~130nm,与纳米粒度仪所测结果是一致的。未氧化的脂质体的形态相对较为规则,大小较集中,边界较清晰,所制得脂质体多为多室脂质体;经Fenton试剂氧化24h后,脂质体形态上与前者出现较大不同,脂质体的粒径接近200nm,并且大小不均,形态不规则,脂质体边界不整齐,处于瓦解边缘。脂质体溶液中出现了脂质体碎片,另外,原来的多室消失,出现了融合的现象。融合是导致脂质体粒径变大的最主要原因。在脂质体氧化96h时,脂质体已经不是球形,脂质体基本完全瓦解崩溃,溶液中出现的是大量的脂质体膜碎片。
2.4 脂质体膜的微极性的变化
计算各胶束体系的I1/I3,根据它随时间的变化绘制得到图4。
根据芘的荧光强度I1/I3值大小可以确定脂质体膜体系的微极性。由于芘元素非极性大,其主要分布在磷脂分子的非极性尾中,即双层膜之间。I1/I3值反映芘周围环境的变化,比值越大,微极性越强,探针所处的环境非极性越弱;比值越小则微极性越弱,探针所处的环境非极性越强。由图4可以看出,随着氧化时间的增加,脂质体膜的微极性大体上略有增加。这是由于磷脂在氧化过程中产生的氧化产物主要是极性小分子,氧化后的磷脂极性增加的趋势,与所预测的是一致的。

图3 脂质体氧化后其形态变化Fig.3 Microscopic image of liposome

图4 脂质体膜微极性的变化Fig.4 The influence of oxidation on liposome micropolaity
2.5 脂质体包封率的变化
测定添加Fenton试剂前后所分离的脂质体荧光强度的变化,进而得到包封率变化。

表2 脂质体包封率的变化Table2 The influence of oxidation on liposomes encapsulation
包埋率可以验证脂质体膜的完整性,钙黄绿素为水溶性物质,因此,钙黄绿素的包埋率越高,膜的结构越致密,膜的完整性程度越高。实验结果表明,脂质体在添加Fenton试剂后,钙黄绿素包封率降低了一倍。磷脂脂肪酸链氧化后发生断裂或基团改变,引起了脂质体膜的降解及融合,使钙黄绿素从脂质体中溶出,从而影响了膜的致密性。将包埋有钙黄绿素的脂质体溶液置于透析袋中,每20h检测一次透析液钙黄绿素荧光强度,结果如图5所示,在加入Fenton试剂50h内,钙黄绿素的泄漏量持续增加;而50h后,透析液中的钙黄绿素含量逐渐下降。实验证实,在氧化初期,脂质体膜发生破裂融合,钙黄绿素在膜降解过程中释放出来,在4d后,脂质体在氧化的作用下基本上全体瓦解,钙黄绿素完全释放出来。

图5 脂质体泄漏率与其氧化时间之关系Fig.5 The relationship between liposome leakage rate and its oxidation time
2.6 脂质体膜的流动性变化
由图6可以看出,经Fenton试剂氧化后,膜的相对流动性下降,膜的流动性主要是依靠脂肪酸的相对运动,氧化后脂肪酸链发生断裂,降解是膜的流动性降低的主要原因。脂质体在添加Fenton试剂后其膜流动性下降,可能是由于氧化后磷脂发生改性,磷脂脂肪酸链可活动自由度降低。
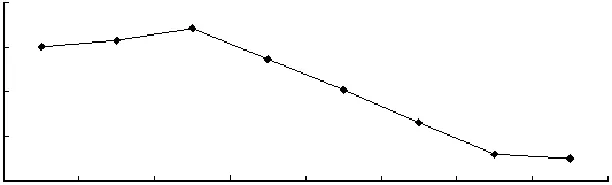
图6 脂质体膜流动性的变化Fig.6 The influence of oxidation on the liposomal membrane relative mobility
3 结论
本实验通过一些较新的实验手段对氧化后的脂质体膜进行的分析。通过对照Fenton反应控制氧化前后脂质体性质的变化,包括MDA值、粒径、Zeta、形态、膜微极性、包埋率和膜流动性,进而确定了氧化对脂质体性质的影响。随着氧化的深入,脂质体的粒径分布更加不集中,由于多室脂质体膜融合,其体积稍有变大,脂质体的形态也不规则,最后脂质体发生了降解,脂质体溶液中出现了脂质体碎片。由于氧化过程中产生的极性小分子,脂质体膜的微极性大体上略有增加。氧化后脂质体包埋率下降,在4d后脂质体所包埋的钙黄绿素基本上释放完全。由于氧化后脂肪酸链发生断裂,膜的相对流动性下降。
[1]Malam Y,Loizidou M,Seifalian A M,et al.Liposomes and nanoparticles:nanosized vehicles for drug delivery in cancer[J].Trends in Pharmacological Science,2009,30:592-599.
[2]Borst J W,Visser N V,Kouptsova O,et al.Oxidation of unsaturated phospholipidsin membrane bilayermixturesis accompanied by membrane fluidity changes[J].Biochimica et Biophysica Acta,2000,1:61-73.
[3]Megli F M,Russo L,Sabatini K,et al.Oxidized phospholipids induce phase separation in lipid vesicles[J].FEBS Letters,2005,579:4577-4584.
[4]Megli F M,Sabatini K.EPR studies of phospholipid bilayers after lipoperoxidation 1.Inner molecular order and fluidity gradient[J].Chemistry and Physics of Lipids,2003,125:161-172.
[5]Cwiklik L,Jungwirth P.Massive oxidation of phospholipid membranes leads to pore creation and bilayer disintegration[J].Chemical Physics Letters,2010,486:99-103.
[6]Soobrattee M A,Neergheen V S,Ramma A L,et al.Kinetics and mechanisms of antioxidant activity using the DPPH free radical method[J].Food Science and Technology,1997,30:609-615.
[7]Min B,Nam K C,Ahn D U,et al.Catalytic mechanisms of metmyoglobin on the oxidation of lipids in phospholipid liposome model system[J].Food Chemistry,2010,123:231-236.
[8]Mohsen M,Mady,Mirhane M,et al.Biophysical studies on chitosan- coated liposomes[J].European Biophysics Journal,2009,38:1127-1133.
[9]Hatzi P,Mourtas S,Pavlos G,et al.Integrity of liposomes in presence of cyclodextrins:Effect of liposome type and lipid composition[J].International Journal of Pharmaceutics,2007,333:167-176.
[10]Wrobel D,Ionov M,Gardikis K,et al.Interactions of phosphorus-containing dendrimers with liposomes[J].Biochimica et Biophysica Acta,2011,3:221-226.
[11]王振华,王玉蓉.脂质体表面电荷的研究进展[C].全国中药创新与研究论坛,2009.
[12]邓英杰.脂质体技术[M].北京:人民卫生出版社,2007:242-245.
