甲醇芳构化中催化剂酸性对脱烷基、烷基化和异构化反应的影响
2013-06-23张金贵骞伟中汤效平黄晓凡
张金贵 骞伟中,* 汤效平, 沈 葵 王 彤, 黄晓凡 魏 飞
(1清华大学化工系,北京市绿色反应工程与工艺重点实验室,北京100084;2华电煤业集团有限公司煤化工事业部,北京100031)
1 引言
由于石油资源日益紧张,以来源于煤或天然气的甲醇做原料,利用催化转化的方法制备烃类尤其是芳烃的技术路线越来越受到重视.1-3使用ZSM-5分子筛,负载镓、铜、锌、银等金属,甲醇一次通过转化能够获得50%-70%收率(碳基)的芳烃.4-10生成的轻烃也可以循环转化,继续生成芳烃.传统石油路线可能得到几百种芳烃与非芳组分,需要专门、复杂、高能耗的芳烃抽提工段来分离.而甲醇芳构化一般只得到苯、甲苯、二甲苯、三甲苯和甲基萘等十余种芳烃,且甲基苯类芳烃的质量分数大于90%.9,10与传统路线相比,该过程具有产品易分离、能耗低的特点.考虑到不同种类的芳烃产物(苯、甲苯和二甲苯)用途不同,理解相关催化剂上芳烃的生成机理,以及影响芳烃产品分布的关键因素,以期高选择性地制备某种芳烃,是进一步研究的目标.
虽然甲醇制芳烃过程生产的芳烃种类较简单,但目前学术界对不同芳烃的生成历程仍然没有统一的认识.前期研究中,李文怀等11使用Zn/ZSM-5分子筛,在350°C转化甲醇,发现可能存在着分子量较大的醚类中间体,在强Brönsted(B)酸催化剂上继续裂解为芳烃与烯烃,但没有讨论芳烃分布的影响因素.Kecskemeti等12比较了二甲醚与二乙醚的芳构化特性,二乙醚本身分子中就存在着烃基链,而甲醇或二甲醚没有,因此甲醇或二甲醚生成C2以上烃类物种的步骤可能影响到产物的分布.当以二甲醚为原料时,产物中二甲苯的含量显著大于甲苯与苯.添加MoC后会促进三甲苯的生成,但甲苯的含量更低.Choudhary等13认为丙烯与丁烯芳构化会率先生成二甲苯,然后二甲苯再进行脱烷基或烷基化反应生成别的甲基苯.Inoue等14利用Ag/ZSM-5在430-470°C转化甲醇,得到了30%-40%的二甲苯与三甲苯,而苯与甲苯的总量低于15%.而Bjørgen等15利用瞬态12C/13C同位素甲醇切换进料技术,分析了甲醇转化为芳烃与烯烃的特征,提出可能存在着双催化循环,一个以三甲苯与甲苯为中间体,继而生成乙烯与芳烃;另一个以高碳烷烃为中间体,继而生成丙烯与其它烷烃.Adebajo等16研究不同温度下甲醇芳构化过程中苯的甲基化二次反应,发现在400°C以上该反应非常显著.显然,反应温度、分子筛(不同Si/Al比和不同酸性)、负载金属和甲醇分压的不同,导致了上述研究结果中芳烃产品分布的差异.
本文认为,如果简化甲醇生成芳烃的具体历程,则甲醇在HZSM-5或改性ZSM-5分子筛上发生转化时,可依次生成二甲醚中间体、烃类中间体,再经聚合、环化和脱氢等步骤形成芳环.3,13,17当芳环生成以后,烷基化、脱烷基和异构化等反应的发生会导致最终芳烃分布与初始芳烃分布的差异.ZSM-5分子筛具有三维交叉孔道体系.直孔道(沿[010]晶面、b轴)大小为0.54 nm×0.56 nm,正弦孔道(沿[100]晶面、a轴)大小为0.51 nm×0.55 nm.18孔道大小与二甲苯分子大小相仿,对直接生成苯、甲苯和对二甲苯均不存在过渡态择形限制.但是根据模拟计算,甲醇芳构化中生成甲苯与对二甲苯在热力学上相对有利.19分子筛的这种择形作用和反应热力学决定了芳烃产品的初始分布.而当催化剂孔内生成的对二甲苯扩散到达表面后,会发生异构化反应生成间二甲苯与邻二甲苯,最终三者组成为热力学平衡组成.20-22同时甲苯和二甲苯可继续发生烷基化反应,分别生成二甲苯和三甲苯.值得指出,气相存在的甲醇分子在催化剂表面吸附生成大量甲氧基与甲基,有利于甲醇与芳环的烷基化反应.23而且甲基类芳烃还会在孔内和表面发生脱烷基化反应,脱除侧链烷基.24,25这些二次反应的发生决定了芳烃产品的最终分布.探究甲醇芳构化过程中烷基化、脱烷基和异构化反应的影响因素,尤其是这些反应和催化剂酸性位点的关系,有助于我们对甲醇制芳烃催化剂的设计和改进,以期在保持高芳烃收率的同时得到理想的芳烃产品分布.
为了探究甲醇芳构化过程中影响主次反应和芳烃分布的因素,本文制备了一系列不同母体Si/Al比和不同Zn负载量的Zn/P/ZSM-5催化剂,在3%Zn/P/ZSM-5(14)催化剂上,分别进行甲醇、二甲苯和甲苯甲醇混合物的转化反应,对应的主反应分别为芳构化、脱烷基和异构化、烷基化反应.芳构化、烷基化和脱烷基反应随着催化剂反应积碳呈现出不同的变化规律.同时结合多种酸性表征手段,探讨不同Si/Al比和Zn含量的催化剂上芳烃产品的分布规律.
2 实验部分
不同Si/Al比的氢型ZSM-5分子筛由南开催化剂厂购得.催化剂体相Si/Al比由电感耦合等离子发射光谱仪(ICP,美国VARIAN公司Vista-MPX型)测定.称取一定量的ZSM-5原粉在(NH4)2HPO4溶液中充分搅拌,浸渍4 h,110°C烘干并在500°C空气气氛下焙烧.磷改性量为3%(以P2O5计).随后分散到不同浓度的Zn(NO3)2溶液中,充分搅拌,浸渍4 h,110°C烘干并在550°C空气气氛下焙烧,得到一系列Zn负载量(1.5%、3%和4.5%(质量分数))的Zn/P/ZSM-5.最终得到的催化剂命名为a%Zn/P/ZSM-5(R),其中a%为Zn负载量,R为催化剂母体Si/Al比.
氨吸附程序升温脱附(NH3-TPD)在美国康塔公司ChemBET Pulsar上进行.将50 mg催化剂样品装入测试装置内,升温至600°C脱除催化剂上吸附的气体物质,降温至100°C,吸附氨气30 min,并使用He气吹出催化剂表面的物理吸附的氨气,随后以10°C·min-1速率进行程序升温并在反应器出口位置检测氨气的浓度.最后,通过标定单位体积氨气的信号值来确定ZSM-5分子筛不同酸性位点的酸密度.
ZSM-5催化剂中B酸和Lewis酸(L酸)的分析使用吡啶吸附的红外光谱法,得到的酸量为半定量结果.仪器型号为Nicolet Nexus 670,工作区域为800-4000 cm-1,分辨率4 cm-1.B酸特征峰在1545 cm-1附近,L酸特征峰在1455 cm-1附近.使用1860和1990 cm-1附近的分子筛特征峰归一化.26B酸密度=1545 cm-1峰面积/(1860 cm-1特征峰面积+1920 cm-1特征峰面积),L酸量=1455 cm-1峰面积/(1860 cm-1特征峰面积+1920 cm-1特征峰面积).取0.25 g催化剂压成直径13 mm的圆片,置于真空池中,450 °C、10-4Pa下脱气4 h,降温到100 °C,吸附吡啶分子1 h,升温到200°C并抽真空到10-4Pa以下,2 h后谱图不再变化时采集谱图,继续升温到350°C并抽真空到10-4Pa以下,2 h后采集谱图.
催化剂的反应评价在微型反应装置中进行.将0.2 g催化剂置于直径10 mm的固定床微反中,将纯甲醇通过预热器预热至200°C,通入反应器中在475°C下进行反应,控制甲醇的质量空速为0.79 h-1.二甲苯或甲苯甲醇混合物进料时过程类似.所得气体与液体产品用气相色谱(日本岛津公司,GC2014)进行分析.芳烃收率是指产物中的总芳烃基于原料的烃基收率,即不考虑甲醇中的水分子.芳烃产品的归一化分布由苯、甲苯、二甲苯或三甲苯的收率除以总芳烃收率后得到.
3 结果与讨论
3.1 不同进料在3%Zn/P/ZSM-5(14)催化剂上的转化反应
积碳改性是ZSM-5常用的修饰手段.一般认为,甲醇反应在催化剂上的积碳分布在外表面和孔道内部,导致酸性中心部分被覆盖.27红外和化学吸附表明,对不同强度的酸性位点,催化剂积碳都会引起酸密度的大量下降.28
在3%Zn改性,母体Si/Al为14的3%Zn/P/ZSM-5(14)催化剂上进行甲醇转化反应,随着反应时间的延长,催化剂上积碳量不断增加.通过分析积碳过程中各芳烃组分收率变化,考察积碳对甲醇制芳烃体系主次反应的影响.
图1为甲醇转化过程中芳烃收率.如图1所示,甲醇一次通过催化剂转化,芳烃碳基收率约为75%左右,包括苯、甲苯、二甲苯、三甲苯和少量乙苯.在142 min的反应时间内,总芳烃收率比较稳定,说明该催化剂芳构化的能力在积碳情况下没有明显降低.而另一方面,各芳烃组分的收率随着积碳增加却呈现规律性变化.苯和甲苯的收率随反应进行逐渐降低,由15 min时的5.7%和21.2%下降到142 min时的2.1%和9.5%.二甲苯的总收率基本不变,同时对、间、邻三个异构体的组成也保持在热力学平衡组成附近,即m(对位):m(间位):m(邻位)=23:53:24.三甲苯的收率随反应进行增加,由15 min时的13.1%增加到142 min时的27.2%.苯和甲苯收率的下降,以及三甲苯收率的提高,说明反应积碳后,脱烷基反应(减少芳环甲基侧链数目)部分被抑制,而烷基化反应(增加芳环甲基侧链数目)则大量发生.为了验证这一点,在类似的反应条件下(475°C、0.1 MPa、2 mL/进料量),换用不同进料进行转化反应,得到原料转化率和产品分布结果见图2和图3.
在甲苯和甲醇同时进料下,甲醇除和甲苯发生烷基化反应生成二甲苯外,还可以单独发生芳构化反应,生成C6-C9芳烃以及C1-C5烯烃和烷烃.二甲苯也可以与甲醇进一步烷基化生成三甲苯.从图2中可以看到,甲醇转化率基本保持在100%,而甲苯转化率则逐渐增加,由15 min时的47.8%增加到75 min时的59.1%.在产品分布中,低碳烃类占比随反应积碳增加逐渐下降,由15 min时的26.3%降低到75 min时的5.8%.其中以丙烷的减少最为明显,由15 min时的13.4%降低到75 min时的1.9%.在此进料体系下低碳烃类,尤其是丙烷的生成,主要来源于甲醇的芳构化反应.该结果表明在大量甲苯存在的情况下,甲醇芳构化反应受到抑制.并且随着反应积碳的增加,芳构化能力变差.同时三甲苯的含量随着反应进行不断增加,说明二甲苯可以继续烷基化生成三甲苯.显然,催化剂积碳量增加,覆盖了进行芳构化的强酸性位点,剩余的弱酸性位点无芳构化能力,但对烷基化反应有利.
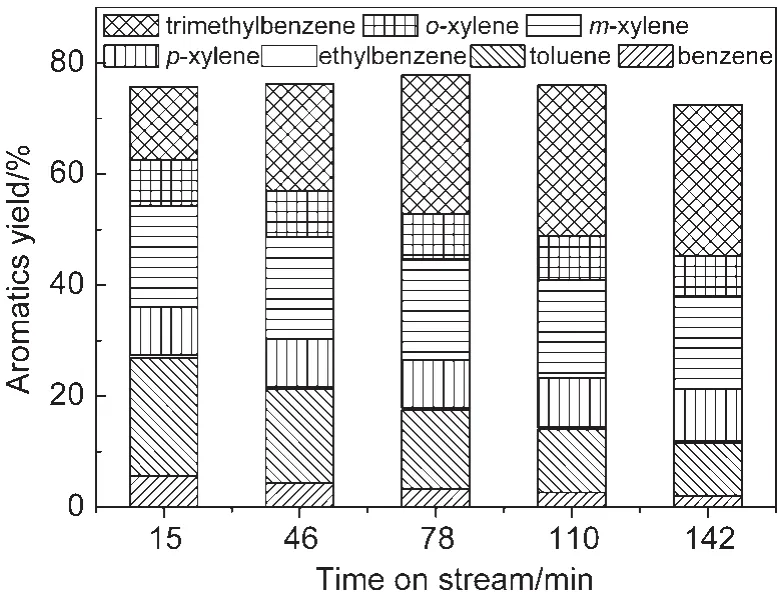
图1 甲醇转化过程中芳烃收率Fig.1 Aromatics yield in methanol conversion reaction
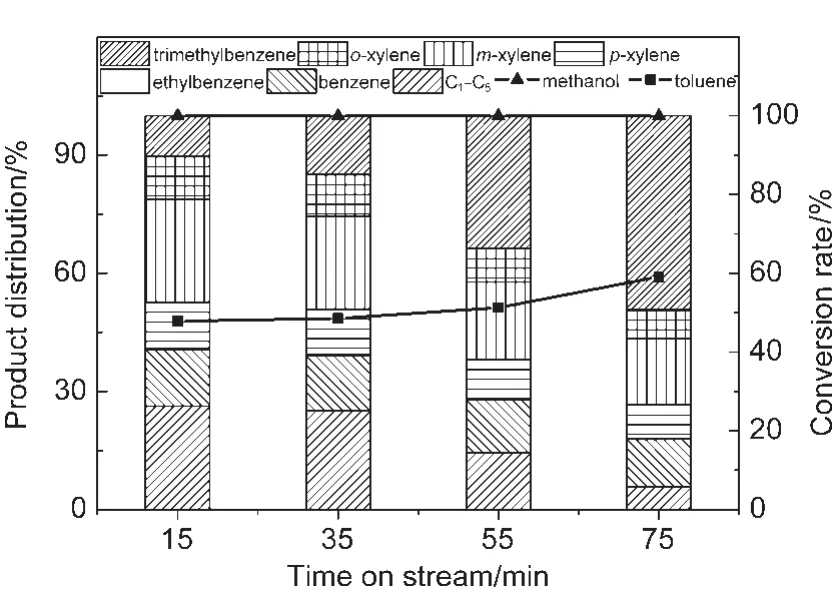
图2 甲苯与甲醇同时进料时甲醇转化率和芳烃产品分布Fig.2 Conversion rates and product distribution under toluene and methanol co-feeding
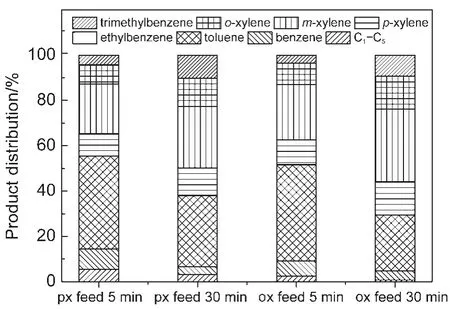
图3 对二甲苯(px)或邻二甲苯(ox)转化的归一化产品分布Fig.3 Normalized aromatics distribution inp-xylene(px)oro-xylene(ox)conversion reaction
在对二甲苯或邻二甲苯进料的情形下,主反应包括:(1)对二甲苯或邻二甲苯异构化得到热力学平衡组成的混二甲苯;(2)二甲苯发生脱烷基反应得到甲苯和苯.另外二甲苯还可能发生歧化和烷基化反应,得到甲苯和少量三甲苯.从图3中可以看到,不管是对二甲苯进料还是邻二甲苯进料,相比反应5 min时,反应30 min时二甲苯通过脱甲基得到的甲苯大大减少.说明催化剂少量积碳对脱烷基反应有强烈的抑制作用.
甲苯与甲醇混合进料和二甲苯进料的实验证明,随着催化剂反应积碳,脱烷基反应被抑制,烷基化反应则加快,这和甲醇进料时的各芳烃收率的变化是吻合的.
总之,催化剂积碳量增加对芳构化、烷基化和脱烷基反应的影响各不相同.在甲醇进料时,芳构化能力不变,而烷基化加快,脱烷基反应被抑制,这也在甲苯甲醇混合进料和二甲苯进料的实验中得到证实.
3.2 不同母体Si/Al比和Zn负载量的Zn/P/ZSM-5催化剂上的甲醇转化反应
反应积碳可以看做是对催化剂酸性的原位改性,通过不同进料下的微反实验可以得到芳构化、烷基化和脱烷基反应在积碳下的变化规律,却难于获得足够的表征结果和反应结果进行关联.为此制备了一系列不同母体Si/Al比和Zn负载量的Zn/P/ZSM-5催化剂,在其上进行甲醇转化反应,并通过表征得到催化剂的酸强度、酸密度和酸性位点类型等性质,探讨催化剂酸性性质对芳构化、烷基化、脱烷基和异构化反应的影响.
选用 Si/Al摩尔比为 14、18.5、24.4和 39.2的HZSM-5催化剂,负载3%的P和3%的Zn,得到一系列3%Zn/P/ZSM-5催化剂.利用NH3-TPD的方法分析催化剂的酸强度和酸密度.
四个催化剂样品的NH3-TPD谱图如图4所示.使用高斯分峰方法进行处理,拟合数据和实验数据能够较好地吻合.谱图中峰的位置代表酸性位点的强弱,峰温越高,酸性越强,从左向右三个峰分别代表弱酸、中强酸和强酸位点.峰的面积代表酸密度的大小,可以利用氨气滴定做外标,得到酸性位点的酸密度.
如图5所示,三种酸性位点的酸密度都随Si/Al比的增加而减少,但减小的幅度有所不同.催化剂Si/Al比由14增加到18.5,强酸、中强酸和弱酸的酸密度大约按照相同的比例减少.Si/Al比由18.5增加到24.4,弱酸酸密度下降放缓,强酸和中强酸酸密度仍然大量减少.Si/Al比由24.4增加到39.2,中强酸酸密度的下降也开始放缓,而强酸酸密度下降趋势不变.Si/Al比为39.2的样品强酸位点密度只有0.006 mmol·g-1,与其他三个样品存在数量级上的差距.
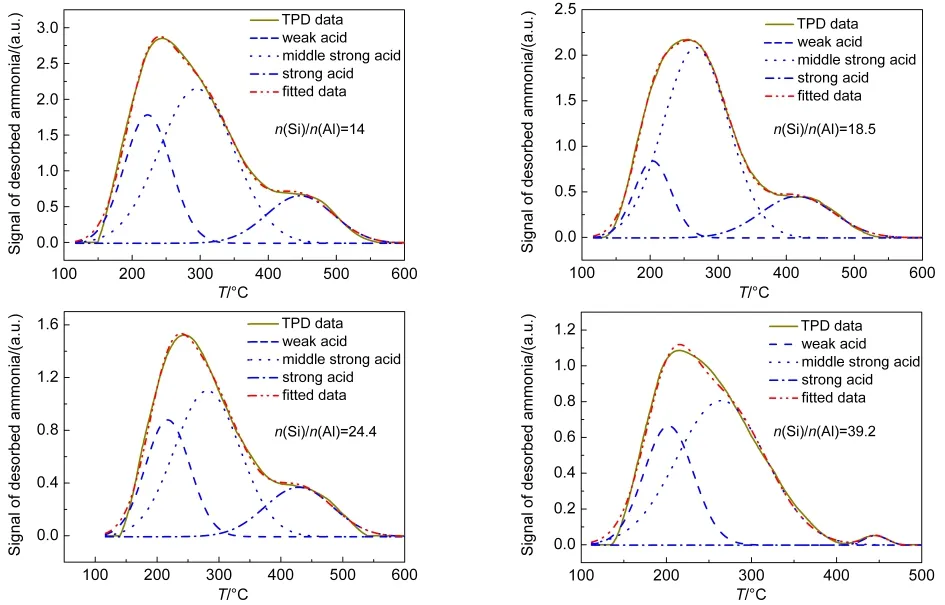
图4 不同n(Si)/n(Al)比的Zn/P/ZSM-5的NH3-TPD谱图Fig.4 NH3-TPD profiles of Zn/P/ZSM-5 with different n(Si)/n(Al)ratios
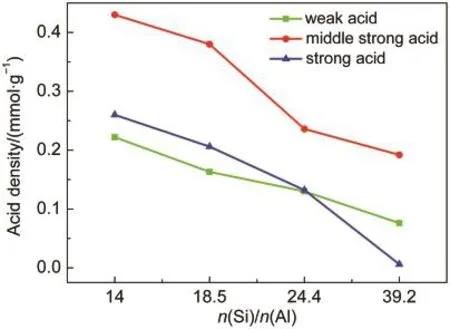
图5 不同n(Si)/n(Al)比Zn/P/ZSM-5催化剂的酸密度Fig.5 Acid density of Zn/P/ZSM-5 catalysts with different n(Si)/n(Al)ratios
从图6可以看到,催化剂Si/Al比由14增加到24.4,催化剂强酸位点酸密度均匀下降,但芳构化性能变化并不大,芳烃收率仅由75.6%下降到73.9%.而归一化的芳烃产品分布却发生了规律性的变化(见图7),表现为苯和甲苯所占比例不断下降,而三甲苯所占比例增加,二甲苯所占比例稍有增加.这与图1中各芳烃组分收率随积碳变化的规律是一致.这说明强酸位点酸密度的下降对脱烷基反应产生了更大影响.而当Si/Al比由24.4增加到39.2时,强酸位点的酸密度大幅度减少,芳烃总收率降低到41.4%,对二甲苯在二甲苯中的选择性由热力学平衡组成的24%增加到45%.二甲苯中对位异构体浓度的提高表明,从孔中扩散出的对二甲苯在外表面的异构化反应得到部分抑制.在强酸位点酸密度下降到一定程度时,芳构化反应和异构化反应都受到抑制,说明二者对催化剂酸强度和酸密度的要求相近.而在强酸位点酸密度超低的催化剂3%Zn/P/ZSM-5(39.2)上,烷基化反应仍在进行,得到大量的二甲苯和三甲苯,说明烷基化反应可在强度较低的酸中心上进行.
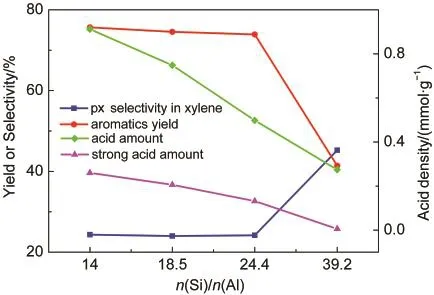
图6 不同Si/Al比的Zn/P/ZSM-5催化剂的酸密度及甲醇反应结果Fig.6 Acid amount and reaction results of Zn/P/ZSM-5 catalysts with different n(Si)/n(Al)ratios
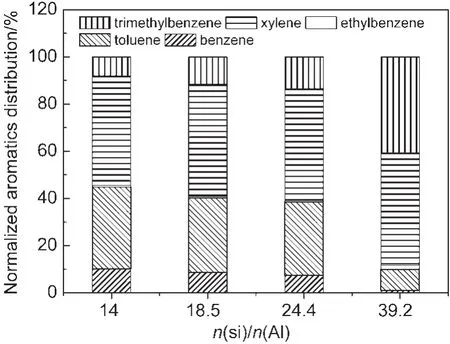
图7 不同n(Si)/n(Al)比催化剂归一化芳烃产品分布Fig.7 Normalized aromatics distribution of Zn/P/ZSM-5 catalysts with different n(Si)/n(Al)ratios
另一方面,为了考察酸性位点类型(B酸或L酸)对甲醇芳构化过程主次反应的影响,制备了一系列不同Zn负载量及母体的Si/Al比为14的Zn/P/ZSM-5(14).吡啶吸附红外谱图如图8所示,其中1455 cm-1处的峰为吡啶与L酸结合的特征峰,1545 cm-1峰为吡啶与B酸结合的特征峰.图中的实线是200°C下脱附吡啶后测量的红外谱图,可以代表催化剂强酸和弱酸的总和,虚线是350°C下脱附吡啶后测量的红外谱图,代表了催化剂的强B酸和强L酸的总和.分别将1455和1545 cm-1峰面积进行归一化处理,代表L酸和B酸的酸量.这种半定量的处理方法适用于本文的讨论各种酸性位点的比例.
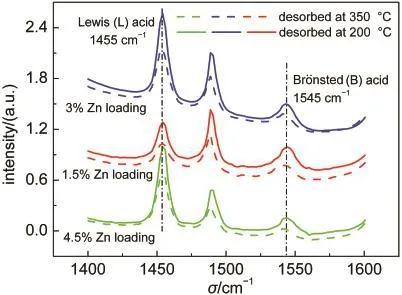
图8 不同Zn负载量的Zn/P/ZSM-5催化剂吡啶吸附红外谱图Fig.8 Py-IR spectra of Zn/P/ZSM-5 catalysts with different Zn loadings
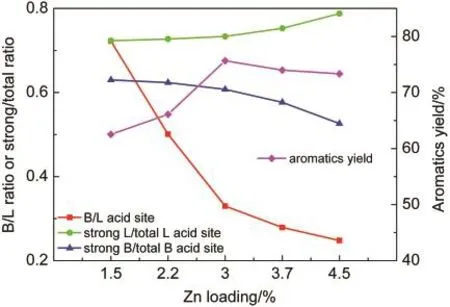
图9 不同Zn负载量催化剂的酸性位点比例和甲醇反应芳烃收率Fig.9 Aromatics yield and acid site ratios of Zn/P/ZSM-5 catalysts with different Zn loadings
随着Zn负载量由1.5%增加到4.5%,L酸峰面积逐渐增大,B酸峰面积逐渐减小,B酸/L酸比下降.这是因为Zn的引入增加了L酸位点,减少了B酸位点.29,30如图9所示,随着Zn负载量的增加和B酸/L酸比例的下降,总芳烃收率呈现出先增大后减少的趋势,在Zn负载量为3%时达到最大.这是因为芳构化是一个B酸与L酸协同作用的催化过程,31过高的或过低的B/L酸比例都对提高芳烃收率不利.
从图9中也看出,Zn改性量对L酸和B酸的酸强度有一定的影响.Zn改性量由1.5%增加到4.5%,强Lewis酸占总Lewis酸的比例由72%增加到79%.而相反的是,强B酸的总B酸的比例由63%降低到53%.B强酸比例的下降影响了芳烃归一化产品的分布.如图10所示,随着Zn负载量的增加,芳烃产品中苯和甲苯占比持续减少,三甲苯占比持续增加.这与图1和图7中芳烃分布的变化规律是类似的,说明了酸强度的降低不利于脱烷基反应,但不影响烷基化反应.
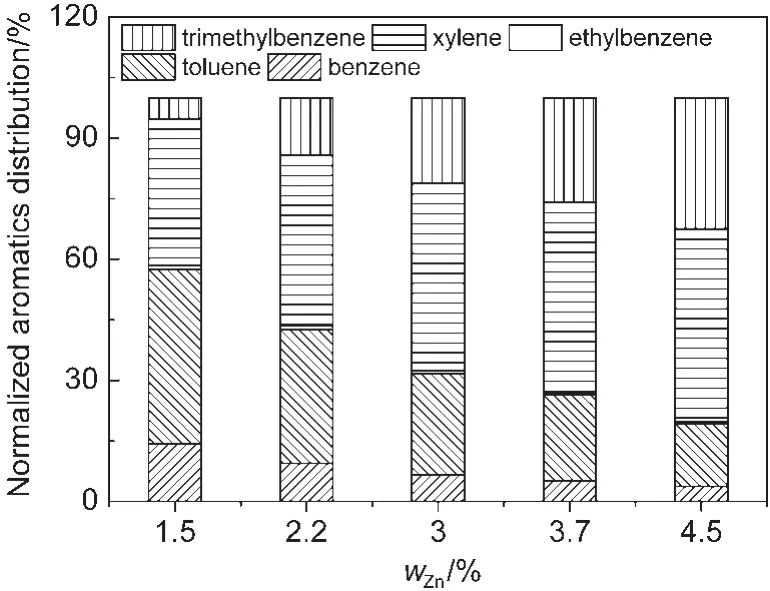
图10 不同Zn负载量的Zn/P/ZSM-5催化剂所得归一化芳烃产品分布Fig.10 Normalized aromatics distribution of Zn/P/ZSM-5 catalysts with different zinc loadings
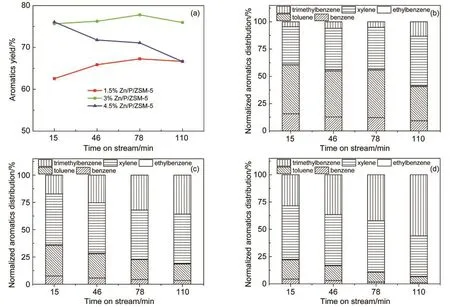
图11 芳烃收率和产品分布随反应时间的变化Fig.11 Aromatics yield and product distribution of Zn/P/ZSM-5 catalysts
进一步考察1.5%、3%和4.5%Zn改性的催化剂上芳烃收率和芳烃产品分布随反应积碳的变化.如图11所示,1.5%Zn改性的催化剂随反应时间延长芳烃收率不断增加,这可能是因1.5%Zn改性的催化剂本身具有过高的B酸/L酸比例,反应积碳使B酸/L酸比例下降,有利于芳烃收率的提高.而4.5%Zn改性的催化剂本身具有过低的B酸/L酸比例,表现出了恰好相反的规律,随反应时间延长,芳烃收率不断下降.3%Zn改性的催化剂芳烃收率基本不变,与图1相比,这两个实验的催化剂改性方法相同但批次不同,结果是类似的.而在芳烃产品分布上,不管Zn改性量的多少,苯和甲苯所占比例都随反应时间延长而下降,说明在不同程度上脱烷基反应都得到了抑制.1.5%Zn改性的催化剂上二甲苯和三甲苯占比都随反应时间延长而增加;3%Zn改性的催化剂上二甲苯占比随反应时间延长基本不变,三甲苯占比随反应时间延长增加;4.5%Zn改性的催化剂上二甲苯占比随反应时间延长下降,三甲苯所占比例随反应时间延长增加.这说明催化剂强酸位点酸密度不同,烷基化反应进行的程度也不同.强酸位点数目越少,越容易发生彻底的烷基化生成三甲苯.强酸位点数目过高,烷基化反应不充分,二甲苯收率不高.3%Zn改性的催化剂强酸位点数目合适,因此具有较高且稳定的二甲苯收率.
选用母体Si/Al比为14、3%Zn负载量的Zn/P/ZSM-5进行甲醇转化,在100%的转化率下能够得到75%的初始芳烃收率,经过两个多小时的反应积碳后,芳构化能力基本不变.二甲苯收率高达35%,且不随积碳增加而发生变化.该催化剂具有良好的工业应用前景.
4 结论
(1)制备的Zn/P/ZSM-5系列催化剂具有良好的芳构化性能.其中母体Si/Al比为14、3%Zn改性的催化剂具有较高的强酸位点酸密度和合适的B酸/L酸比例,甲醇转化的芳烃收率高达75%.
(2)随着催化剂反应积碳,芳构化能力基本不变,而脱烷基反应受到抑制,烷基化反应在严重积碳后仍然进行.
(3)不同Si/Al比和Zn负载量催化剂上的甲醇转化反应表明,芳构化、脱烷基和烷基化反应对强酸位点酸密度要求不同.随着强酸位点酸密度下降.首先受到抑制的是脱烷基反应,其次是芳构化反应,而烷基化反应在强酸密度很低的情况下仍能进行,并且将生成大量三甲苯.控制强酸位点酸密度可以有效地控制芳烃产品分布.
(4)芳构化和异构化反应对强酸位点酸密度的要求相似,因此,在消除酸性位点来提高对位选择性的同时往往会带来芳烃收率的下降.考虑到异构化反应基本在外表面进行,为了尽可能减少对芳构化反应的影响,选择性消除外表面酸性位点是抑制异构化反应的一个思路.
(1) Olsbye,U.;Svelle,S.;Bjorgen,M.;Beato,P.;Janssens,T.V.W.;Joensen,F.;Bordiga,S.;Lillerud,K.P.Angew.Chem.Int.Edit.2012,51,2.doi:10.1002/anie.201107584
(2) Keil,F.J.Microporous Mesoporous Mat.1999,29,49.doi:10.1016/S1387-1811(98)00320-5
(3) Stocker,M.Microporous Mesoporous Mat.1999,29,3.doi:10.1016/S1387-1811(98)00319-9
(4) Zaidi,H.A.;Pant,K.K.Catal.Today2004,96,155.doi:10.1016/j.cattod.2004.06.123
(5) Freeman,D.;Wells,R.P.K.;Hutchings,G.J.J.Catal.2002,205,358.doi:10.1006/jcat.2001.3446
(6)Freeman,D.;Wells,R.P.K.;Hutchings,G.J.Chem.Commun.2001,1754.
(7)Ni,Y.M.;Peng,W.Y.;Sun,A.M.;Mao,W.L.;Hu,J.L.;Li,T.;Li,G.X.J.Ind.Eng.Chem.2010,16,503.doi:10.1016/j.jiec.2010.03.011
(8) Lopez-Sanchez,J.A.;Conte,M.;Landon,P.;Zhou,W.;Bartley,J.K.;Taylor,S.H.;Carley,A.F.;Kiely,C.J.;Khalid,K.;Hutchings,G.J.Catal.Lett.2012,142,1049.doi:10.1007/s10562-012-0869-2
(9)Tian,T.;Qian,W.Z.;Sun,Y.J.;Cui,Y.;Lu,Y.Y.;Wei,F.Modern Chemical Industry2009,29,55.[田 涛,骞伟中,孙玉建,崔 宇,卢俨俨,魏 飞.现代化工,2009,29,55.]
(10)Tian,T.;Qian,W.Z.;Tang,X.P.;Yun,S.;Wei,F.ActaPhys.-Chim.Sin.2010,26,3305.[田 涛,骞伟中,汤效平,恽 松,魏 飞.物理化学学报,2010,26,3305.]doi:10.3866/PKU.WHXB20101228
(11)Wang,J.Y.;Li,W.H.;Hu,J.X.Journal of Fuel Chemistry and Technology2009,37,607.[王金英,李文怀,胡津仙.燃料化学学报,2009,37,607.]
(12) Kecskemeti,A.;Barthos,R.;Solymosi,F.J.Catal.2008,258,111.doi:10.1016/j.jcat.2008.06.003
(13) Choudhary,V.R.;Panjala,D.;Banerjee,S.Appl.Catal.A2002,231,243.doi:10.1016/S0926-860X(02)00061-3
(14)Inoue,Y.;Nakashiro,K.;Ono,Y.Microporous Mat.1995,4,379.doi:10.1016/0927-6513(95)00020-A
(15) Bjørgen,M.;Svelle,S.;Joensen,F.;Nerlov,J.;Kolboe,S.;Bonino,F.;Palumbo,L.;Bordiga,S.;Olsbye,U.J.Catal.2007,249,195.doi:10.1016/j.jcat.2007.04.006
(16)Adebajo,M.O.;Long,M.A.Catal.Commun.2003,4,71.doi:10.1016/S1566-7367(02)00259-5
(17)Lukyanov,D.B.;Gnep,N.S.;Guisnet,M.R.Ind.Eng.Chem.Res.1995,34,516.doi:10.1021/ie00041a012
(18)Olson,D.H.;Kokotailo,G.T.;Lawton,S.L.;Meler,W.M.J.Phys.Chem.1981,85,2238.doi:10.1021/j150615a020
(19)Joshi,Y.V.;Thomson,K.T.J.Phys.Chem.C2008,112,12825.doi:10.1021/jp712071k
(20) Olson,D.H.;Haag,W.O.ACS Symp.Ser.1984,248,275.doi:10.1021/symposium
(21) Mirth,G.;Cejka,J.;Lercher,J.A.J.Catal.1993,139,24.doi:10.1006/jcat.1993.1003
(22)Guisnet,M.;Gnep,N.S.;Morin,S.Microporous Mesoporous Mat.2000,35-36,47.
(23) Ivanova,I.I.;Corma,A.J.Phys.Chem.B1997,101,547.doi:10.1021/jp961468k
(24) Tsai,T.;Liu,S.;Wang,I.Appl.Catal.A1999,181,355.doi:10.1016/S0926-860X(98)00396-2
(25) Serra,J.M.;Guillon,E.;Corma,A.J.Catal.2004,227,459.doi:10.1016/j.jcat.2004.08.006
(26) Zheng,S.R.;Heydenrych,H.R.;Jentys,A.;Lercher,J.A.J.Phys.Chem.B2002,106,9552.doi:10.1021/jp014091d
(27) Bibby,D.M.;Howe,R.F.;Mclellan,G.D.Appl.Catal.A1992,93,1.doi:10.1016/0926-860X(92)80291-J
(28)Benito,P.L.;Gayubo,A.G.;Aguayo,A.T.;Olazar,M.;Bilbao,J.Ind.Eng.Chem.Res.1996,35,3991.doi:10.1021/ie950462z
(29) Biscardi,J.A.;Meitzner,G.D.;Iglesia,E.J.Catal.1998,179,192.doi:10.1006/jcat.1998.2177
(30) El-Malki,E.M.;Van Santen,R.A.;Sachtler,W.M.H.J.Phys.Chem.B1999,103,4611.doi:10.1021/jp990116l
(31) Bhan,A.;Delgass,W.N.Catal.Rev.2008,50,19.doi:10.1080/01614940701804745
