沉水植物生态修复对西湖细菌多样性及群落结构的影响*
2013-05-28李琳琳汤祥明邵克强龚志军张云华
李琳琳,汤祥明,高 光,邵克强,龚志军,陈 丹,张云华
(1:安徽农业大学生命科学学院,合肥 230036)
(2:中国科学院南京地理与湖泊研究所湖泊与环境国家重点实验室,南京 210008)
近年来,湖泊富营养化已成为世界范围内一个突出的环境问题[1-3].随着富营养化的加剧,湖泊中常暴发藻类水华,水生植物急剧减少,甚至消失.而大型水生植物,特别是沉水植物,能吸收利用水体中的营养物质,合成自身生长发育所需要的物质,有效地降低水体中的营养盐浓度,同时与藻类竞争营养盐,抑制“藻华”的暴发.因此,沉水植物的恢复往往是湖泊富营养化修复的主要措施之一[4-6].
在水生态系统中,微生物是极为敏感并易受环境影响的生物类群.它们不仅是系统中生物量的重要组成部分,而且影响着物质循环和营养传递过程[7].鉴于细菌在水生态系统中的重要作用及其对生态系统的变化反应迅速等特点,在生态修复过程中,细菌的丰度、群落结构及多样性等的变化可以作为评判生态修复效果的重要指标[8-9].
杭州西湖是我国著名的风景旅游湖泊,也是一个浅水型城市富营养化湖泊,水域面积5.6 km2,平均水深仅1.56 m.湖水的总氮(TN)和总磷(TP)分别为2.204 和0.125 mg/L,平均透明度仅为46 cm[10].经过一系列整治工程后,水质得到了一定程度的改善,如北里湖TN 和TP 分别为1.930 和0.041 mg/L,但湖泊富营养化问题依然没有解决[11].目前,关于西湖细菌的研究主要集中在对西湖水体及沉积物中可培养细菌方面[10,12-14],但天然水环境中的微生物仅有0.001%~3%可以被分离培养[15],利用传统的平板菌落计数方法不能获取水体中比较真实的细菌丰度信息,更无法了解特定生态修复措施是如何导致生境中微生物种群组成的变化.
近些年,随着分子生物技术的迅速发展,基于原核生物16S rRNA 基因的聚合酶链式反应-变性梯度凝胶电泳(PCR-DGGE)及克隆文库等技术,已广泛应用于湖泊生态系统的微生物群落结构研究[16-21].湖泊生态修复研究表明,沉水植物可显著改变水体中细菌的多样性[22-23],但沉水植物对水体中细菌群落组成的影响却少有报导.本文采用PCR-DGGE 和克隆文库相结合的方法,研究了杭州西湖沉水植物生态修复对水体细菌多样性及群落结构的影响,以期为富营养化湖泊生态系统修复机制提供理论依据.
1 材料和方法
1.1 实验设计及采样地点
为了改善西湖水质,实施了以沉水植被恢复为主的生态修复工程.实验围隔建在西湖北面的北里湖(30°15'26″N,120°8'16″E;图 1),由聚乙烯布围成,共 6 个,每个围隔 6 m 长、4 m 宽、1.7 m 深.围隔底部先铺上竹排再加上西湖底泥,厚约40 cm.2011年1月在1#~3#围隔种植菹草(Potamogeton crispus),4#~6#围隔撒播苦草(Vallisneria natans)块茎(图1).本次试验采样点分别设在6 个围隔(1#~3#围隔记作菹草处理;4#~6#围隔记作苦草处理)及围隔外北里湖未进行生态修复的湖区7#~9#(记作无草对照).4月底采样时1#~3#围隔菹草繁茂,株高约1.2 m,覆盖度达到100%;4#~6#围隔苦草长出约15 cm,覆盖度约为50%.
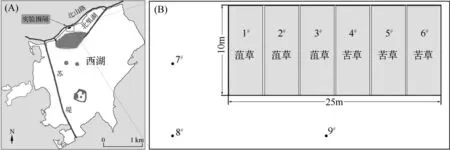
图1 杭州西湖生态修复工程位置、围隔设置及采样点Fig.1 Location of ecological restoration project,design of enclosures and sampling sites in West Lake,Hangzhou
1.2 样品的采集及处理
于2011年4月29日,用采水器采集每个围隔内及对照点的表层水样(水下约50 cm 处)各5 L.水样分成3 个部分:(1)取250 ml 水样,现场用手持式真空泵于0.2 μm 孔径的聚碳酸酯滤膜上过滤,然后用灭过菌的离心管收集滤膜,置于-20℃保存,一周内进行DNA 提取及细菌群落结构分析.(2)另取46 ml水样装入灭过菌的离心管中,管中预先加入了用0.2 μm 孔径的滤膜过滤后的无颗粒甲醛溶液4 ml(甲醛的终浓度为2%V/V,体积比),样品置冰盒中运回实验室后置冰箱4℃保存,一周内进行细菌丰度测定.(3)其余水样置于预先洗净的塑料桶中送到实验室直接用于水样化学及生物指标分析.
1.3 理化参数的测定
采样点水体的透明度(SD)用透明度盘现场测定.水深用便携式测深仪(奥地利UWITEC 公司)现场测定.水温、pH、溶解氧(DO)、浊度及叶绿素a(Chl.a)的含量用多参数水质测定仪(YSI6600V2,USA)现场测量.总磷、总氮、溶解性总氮(DTN)、溶解性总磷(DTP)含量的分析参照《湖泊富营养化调查规范》[24].悬浮物含量(SS)为水样经Whatman GF/F 膜过滤后将滤膜在105℃烘干4 h 至恒重后的质量[24];称重后的滤膜放入马弗炉,在550℃下灼烧3 h 至恒重后再次称重,两次质量之差即为悬浮物中有机质的含量.
1.4 细菌计数
采用DAPI 染色后表面荧光显微镜直接计数的方法[25].将上述甲醛固定的水样用无菌水稀释一定倍数,用核酸染料DAPI 染色,于表面荧光显微镜在16×100 放大倍数下观察计数,每个样品不少于20个视野,再将视野里的细菌个数转换成每毫升细菌数,即为细菌丰度.
1.5 DNA提取及PCR-DGGE分析
水样细菌总基因组DNA 的提取参照Zhou 等的方法[26].采用针对细菌16S rDNA 的特异性引物进行PCR 扩增,正向引物是341f(5'-CCTACGGGAGGCAGCAG-3'),引物的5'端连有一个40 bp 的GC 夹;反向引物为 518 r(5'-ATTACCGCGGCTGCTGG-3')[16].PCR 采用 25 μl 反应体系,组份为:10× PCR 缓冲液,25 mmol/L MgCl2,2.5 mmol/L dNTP,0.5 μmol/L 引物,1.0 U Taq 酶(Takara)和 1 μl 模板 DNA,加无菌水补足至25 μl.PCR 扩增反应在ABI 公司的热循环仪(Applied Biosystems VeritiTMThermal Cycler)中进行,采用降落PCR 扩增程序:94℃预变性5 min,94℃变性30 s,65 ~53℃(每个循环温度下降1℃)退火30 s,72℃延伸30 s,13 个循环;采用53℃的退火温度再进行21 个循环;最后72℃延伸7 min.PCR 结束后进行琼脂糖凝胶电泳,确定片段大小,用图像分析软件拍照分析.
变性梯度凝胶电泳使用美国CBS 公司电泳仪(DGGE-2001)进行,使用8%的聚丙烯酰胺变性胶(丙稀酰胺∶双丙稀酰胺=37.5∶1),变性梯度为40%~60%(100%变性胶含有7 mol/L 尿素和40%去离子甲酰胺).电泳缓冲液为1× TAE(20 mmol/L Tris,10 mmol/L acetate,0.5 mmol/L EDTA,pH 8.0).60℃恒压100 V,电泳16 h.DGGE 胶用SYBR GREEN I(1∶10000 稀释)染色15 ~30 min,采用Omega10TM全自动多功能凝胶成像分析系统拍照.
1.6 样品的克隆建库
根据DGGE 结果,选择1#、4#、7#样品克隆建库,使用细菌通用引物27f(5'-AGAGTTTGATCMTGGCTCAG-3')和 1492 r(5'-GGTTACCTTGTTACGACTT-3')扩增细菌的 16S rDNA.反应体系(25 μl):2.5 μl 10× buffer,0.25 μl 模板 DNA,2 μl dNTP,各 0.3 μl 的引物和 0.4 μl Taq 聚合酶,无菌水补至 25 μl.使用降落 PCR 进行扩增,程序如下:94℃预变性 5 min,94℃变性45 s,65 ~52℃(每循环下降0.5℃)退火45 s,72℃延伸90 s,24个循环;采用52℃的退火温度再进行6 个循环;最后72℃延伸7 min.将3 次PCR 产物混合后通过1%琼脂糖凝胶电泳,割胶后纯化,立即进行TA 克隆,将纯化后的PCR 产物连接到pGEM-T Easy 载体(Promega)上,然后转化入大肠杆菌Top 10 感受态细胞,构建克隆文库.
1.7 测序和系统进化树分析
每个文库随机挑选100 个阳性克隆进行测序.测序由上海美吉生物医药科技有限公司完成.所得序列使用BioEdit(version 7.0.9)剪切留取置信度最高的750bp 序列.利用RDP 在线程序Check-Chimera 对获得的16S rDNA 序列片段进行嵌合体(Chimera)检验,并通过BLAST(http://www.ncbi.nlm.nih.gov/blast)进行数据库比对,去除嵌合体和叶绿体(Chloroplast)序列.将相似性高于97%的序列归为同一个操作分类单元(operational taxonomic unit,OTU),每一个OTU 中选择代表性克隆子(3 个文库中各选一个)在NCBI 数据库中进行BLAST 比对,选择参比序列,然后使用MEGA 5.0 (Molecular Evolutionary Genetics Analysis,version 5.0)软件,根据Jukes-Cantor 算法计算进化距离,用最小进化距离法(ME 法)构建系统进化树.进化树拓扑学分析的置信值由1000 次随机取样后,经最大简约分析(maximum parsimony analysis)计算所得.本研究所得到的248 个细菌16S rDNA 序列已上传到GenBank 数据库,登录号为JX075268 ~JX075515.
1.8 数据处理
所得到的数据用统计软件SPSS 17.0 进行单因素方差分析(One way ANOVA),以比较不同沉水植物生态修复与无草对照点间差异的显著性.DGGE 结果用GelCompar II 软件(Applied Maths,Kortrijk,Belgium)进行分析,以获得DGGE 条带信息的矩阵,计算各样品细菌群落的多样性指数,并进行聚类分析.克隆文库覆盖度及多样性指数等用软件SPADE(Chao and Shen,http://chao.stat.nthu.edu.tw/softwareCE.html)进行计算.文库差异的显著性通过Mothur 软件(版本v1.24.0)中的∫-Libshuff 程序进行比较[27].
2 结果
2.1 水质参数
菹草处理、苦草处理与湖区无草对照之间pH 无显著差异,均值为9.01;无草对照点平均水温为23.47℃,分别比菹草处理和苦草处理低0.70 和1.40℃(图2A).种植沉水植物显著降低了水体的浊度和SS含量,使水体的透明度提高了82%,但显著增加了水体中有机质的比例(图2B、C).种植沉水植物也显著降低了水体中TP、TN 及DTN 的浓度,但对水体中DTP 的影响不大(图2D、E).种植菹草和苦草均使水体中DO 浓度升高,但与无草对照组之间差异不显著(图2F).沉水植物的恢复显著降低了水体中的Chl.a 的含量(图2G).
2.2 细菌丰度
北里湖无草对照点细菌丰度均值为2.96×106cells/ml,种植菹草和苦草使细菌丰度分别降至2.27×106和2.51×106cells/ml,但三者间的差异未达到显著水平(图2H).
2.3 细菌PCR-DGGE分析
DGGE 结果显示无草对照DGGE 条带数均值为30 条,菹草处理和苦草处理的DGGE 条带数均值分别为40 条和34 条,且菹草处理与无草对照相比差异显著(图2I).聚类分析表明在相似度为70%的水平上,无草对照、菹草处理和苦草处理细菌群落分别形成3 个明显不同的类群(图3).根据DGGE 结果计算出来的细菌多样性指数也表明:种植菹草和苦草可显著提高水体中细菌的Shannon 指数(图2J).
2.4 克隆文库的分析
3 个细菌16S rDNA 克隆文库去除嵌合体序列及28 个来自硅藻门的Chloroplast 序列(均来自菹草处理的文库)后,共得到248 个有效序列,44 个OTU(表1).3 个文库的覆盖度在77.8% ~95.6%之间,说明文库的代表性比较高.文库的Chao1 指数数据表明,种植水草后水体中细菌种类的丰富度有所增加,但反映克隆文库中OTU 的丰度、均匀度等的综合指标的Shannon 指数(H')变化趋势不一(表1).∫-Libshuff 结果表明:种植菹草和苦草的处理组之间细菌群落差异不显著,但两者均与无草对照组的细菌群落结构差异显著(表2).
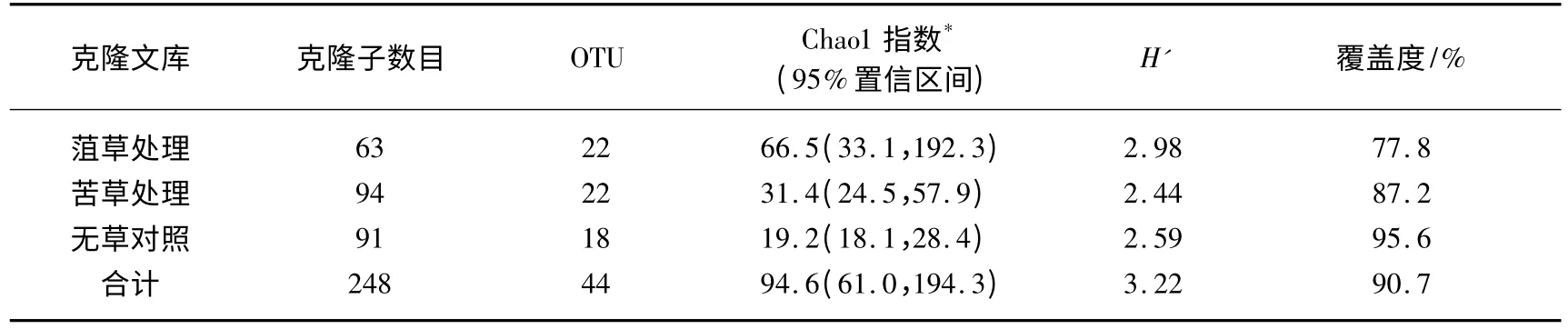
表1 细菌克隆文库多样性比较Tab.1 Comparison of bacterial diversity and library coverage estimations in the clone libraries
2.5 系统发育分析
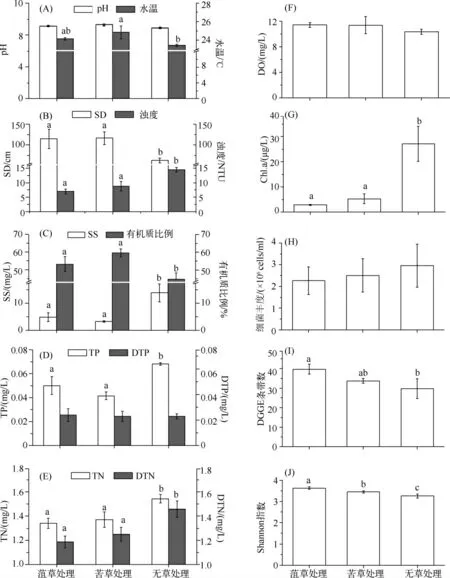
图2 西湖不同处理围隔及湖区无草对照点主要的水质及生物参数比较(数据为均值±标准差;不同字母表示同一指标不同处理下差异显著(P <0.05))Fig.2 Comparison of water quality and biological parameters among two kinds of ecological restorations and the control stations in West Lake
北里湖细菌由拟杆菌门(Bacteroidetes)、疣微菌门(Verrucomicrobia)、放线菌门(Actinobacteria)和变形菌门(Proteobacteria)中的 Alpha-、Beta-及 Gamma-proteobacteria 3 个纲组成.Bacteroidetes、Betaproteobacteria 和Verrucomicrobia 是3 个最主要的细菌门类,分别占到42.9%、30.8%和14.3%(图4).种植菹草和苦草的围隔中Bacteroidetes 所占的比例分别减少了30.2%和37.6%;Betaproteobacteria 所占的比例分别提高了21.6%和28.8%;Alphaproteobacteria 则由4.4%提高到19.0%和12.8%.种植菹草后Verrucomicrobia 的比例大幅降低,而种植苦草对Verrucomicrobia 的影响不大.此外,种植菹草的围隔中Gammaproteobacteria 的比例提高了5.1%,并且浮霉菌门(Planctomycetes)的细菌也只发现存在于这种围隔中.

图3 不同处理围隔中和湖区无草对照点细菌群落组成的DGGE 图谱及聚类分析Fig.3 Bacterial community composition analyzed by DGGE profiles and cluster analysis of the samples
通过与已培养的细菌进行序列相似性比对发现[28],248 序列与已培养的细菌的相似性在82.2%~100%之间,其中相似性大于97%的序列占到50.4%.细菌16S rDNA 的系统发育树表明,3 个文库中共有70.2%(174 个)的序列属于15 个已知的典型淡水细菌类群[29-32],如属于Betaproteobacteria 的 Rhodoferax sp.BAL47、Li-UU-5-340.2、P.necessarius、PRD18A09 等;属于Alphaproteobacteria 的 GOBB3-C201、LiUU-9-283.2等;属于 Bacteroidetes 的 PRD01001B 等;属于Verrucomicrobia 的 CL0-14 等(图 5).其中属于Rhodoferax sp.BAL47、CL0-14、GOBB3-C201 和PRD01001B 这4 个类群的细菌序列最多,分别为 77、30、24 和 22 个.Rhodoferax sp.BAL47 类群的克隆子最相近的菌株是最近分离纯化自捷克Rimov 水库的两个菌株Limnohabitans planktonicus II-D5T和Limnohabitans parvus II-B4T[33],属于其它3 个类群的克隆子没有相似性大于97%的相应菌株.
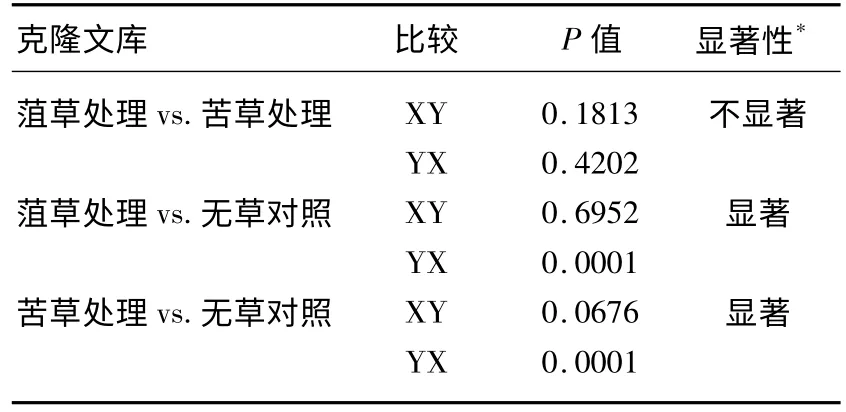
表2 细菌群落结构差异性显著性的∫-Libshuff 分析Tab.2 ∫-Libshuff comparisons of the heterogeneity of the three bacterial clone libraries
3 讨论
3.1 沉水植物生态修复对水体中细菌多样性的影响
氮、磷及有机碳等是细菌增殖的限制因素,通常氮、磷浓度的升高可促进湖泊中细菌的增殖,但水体营养盐的过度升高也会造成沉水植物的消亡、微生物多样性下降等生态环境的恶化,最终导致湖泊生态系统受损[34-37].本研究表明,种植沉水植物后水体中的细菌数量有所下降,但是细菌的DGGE 条带数有所增加,并且表征细菌多样性的Shannon 指数也显著高于无草对照水域(图2),这与毛杰等研究的结果一致[23].
围隔实验发现,进行菹草和苦草生态修复可显著降低水体中TN、TP 浓度及Chl.a 的含量,同时降低水体的浊度及悬浮物的浓度,显著提高水体的透明度(图2),使生态系统由以浮游植物为主的浊水态系统转变为以沉水植物为优势的清水态系统.沉水植物的生长可吸收大量的营养盐,同时抑制沉积物中营养盐的释放,而水体中氮、磷等营养盐的降低可能是导致围隔中细菌丰度降低的主要原因.由于竞争和化感作用,沉水植物可能抑制浮游植物的生长,导致处理组叶绿素含量远低于无草对照组,而浮游植物群落可以显著影响浮游细菌的多样性[30].其次,沉水植物的生长增加了水体微环境的异质性,为细菌提供更加多样的生态位,这可能是生态修复后细菌多样性增加的另一个主要原因.此外,种植沉水植物后水体中有机质比例显著高于湖区无草对照(图2C),这暗示沉水植物降低营养盐和浮游植物生物量的同时,自身释放一定的溶解性有机碳(DOC).而研究表明DOC 对细菌群落及其多样性有一定的调控作用[9,38].
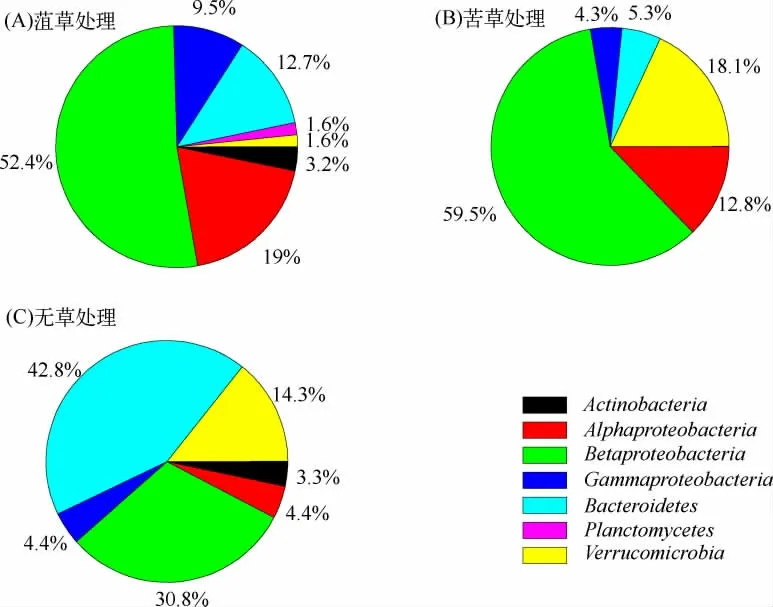
图4 不同沉水植物处理下细菌种类组成的百分比Fig.4 Relative abundance of bacterial phylogenetic groups in the studied clone libraries
3.2 沉水植物生态修复对水体中细菌群落结构的影响
革兰氏阳性菌Bacteroidetes(即以前的Cytophaga-Flexibacter-Bacteroides,CFB)和革兰氏阴性菌α-、β-proteobacteria 是富营养化的淡水湖泊生态系统中最为丰富的细菌菌群.研究表明,沉水植物的存在可显著改变水体中细菌的群落组成[20,39].本实验发现,沉水植物生态修复后水体中的Bacteroidetes 比例大为降低,而α-、β-proteobacteria 比例迅速上升(图4).这种细菌类群比例的变化,主要表现为少数典型淡水类群中克隆子数量的增减和部分新OTU 的产生(图5、6).例如,无草对照点湖水中属于Bacteroidetes 的一个OTU(代表克隆子为:XH-4-B-80,共有21 个克隆子,图5)就占到Bacteroidetes 中克隆子总数的53.8%,与此OTU 最相似的序列clone E4(HQ853002)来自富营养化的洱海.而经沉水植物修复后的水体中此OTU 的细菌几乎消失.Bacteroidetes 数量的减少并伴随α-和γ-proteobacteria 数量的增加可能是水质改善的表现[40-41].
细菌群落结构的变化是其对环境变化的适应,也与其功能相一致.沉水植物修复可通过抑制沉积物再悬浮和抑制浮游植物的过度繁殖、释放可溶性有机物等,改变局域水体的水质,甚至生态系统的类型(浊水型转变为清水型),最终也影响到水体中细菌的多样性、群落组成及生态学功能.例如,湖泊中α-proteobacteria被认为在低营养盐环境中更有竞争力,并且它们有降解复杂有机复合物(可能来自沉水植物的释放或降解)的能力,而Bacteroidetes 常与藻类释放的有机物降解关系密切[30,42].虽然本实验发现菹草和苦草对水体中细菌多样性和细菌群落组成的影响不完全一致(图4、5、6),但是这有可能与实验时这两种植物所处的生长时期和覆盖度不同有关.采样时菹草生长已经达到鼎盛期,覆盖度达到了100%;而此时苦草刚处于生长初期,覆盖度也只有50%左右.不同种类的沉水植物或不同组合的沉水植物对水体中细菌的影响是否相同,仍需要更多的实验数据加以揭示.
综上所述,沉水植物生态恢复不仅可以提高水质,也可以提高水体中细菌的多样性,改变水体中细菌的群落组成,使之向更加健康的方向发展.在控源截污的基础上,进行沉水植物生态恢复与维持,并配以适当的工程措施和管理,可加快湖泊生态修复进程.
致谢:感谢朱广伟研究员、赵林林、薛庆举、杨尚等在采样及水样化学分析中给予的帮助.
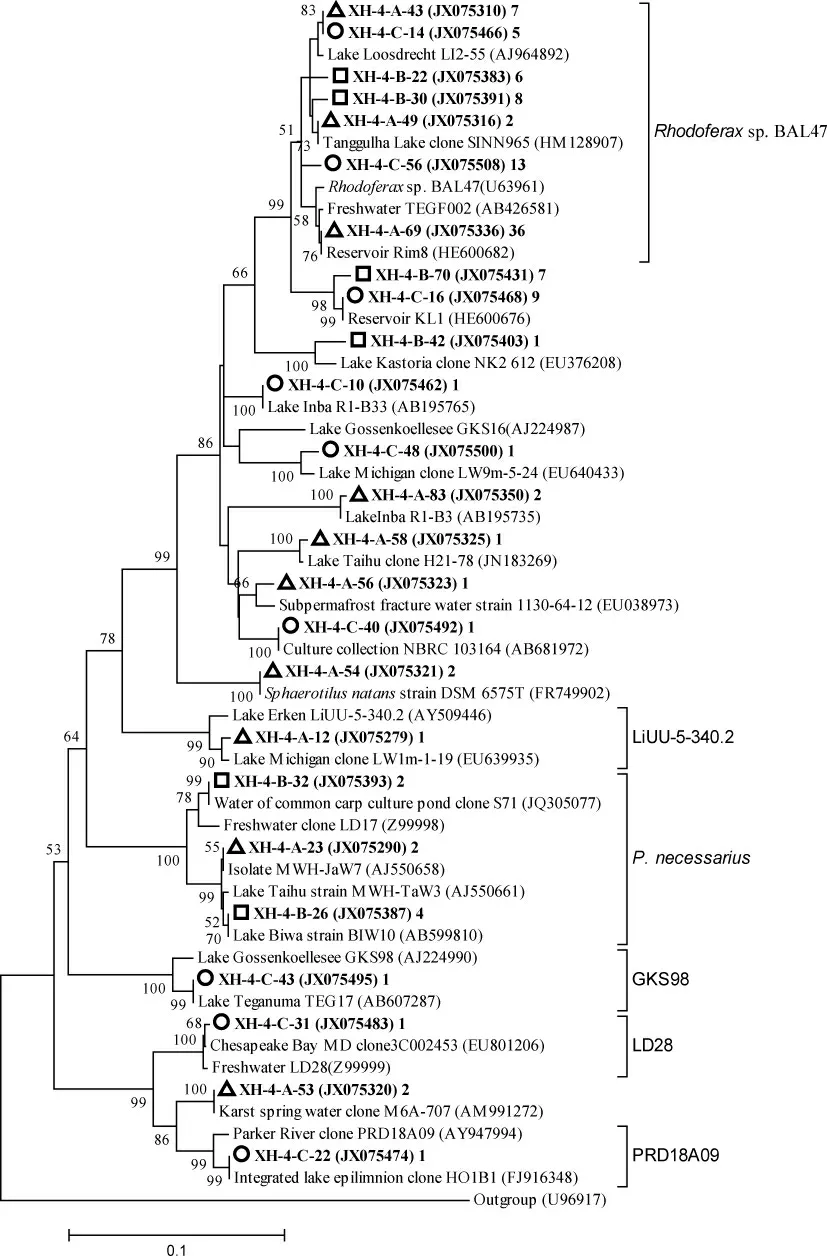
图5 Betaproteobateria 基于16S rDNA 的系统发育树(本研究所获得的克隆子加粗显示.克隆子前面的符号表示不同的文库:菹草处理(○),苦草处理(△),无草对照(□).克隆子后括号内为其GenBank 登录号,最后面为同一文库中属于同一OTU 的克隆子数目.从GenBank 数据库获得的相似序列的生境置于其登录号之前.中括号后为典型淡水浮游细菌类群,下同)Fig.5 Minimum evolution trees inferred by analysis of partial 16S rDNA sequences obtained from the tree studied clone libraries shown for Betaproteobacteria (The numbers of clones in each OTU (shown as bold)are given in parentheses.The symbols before each OTU donate its origin:Potamogeton crispus treatment (○),Vallisneria natans treatment (△)and the control (□).The habitats of the most related sequences are given before their GenBank accession numbers.The reported typical freshwater bacterial clusters are shown after corresponding brackets,the same below)
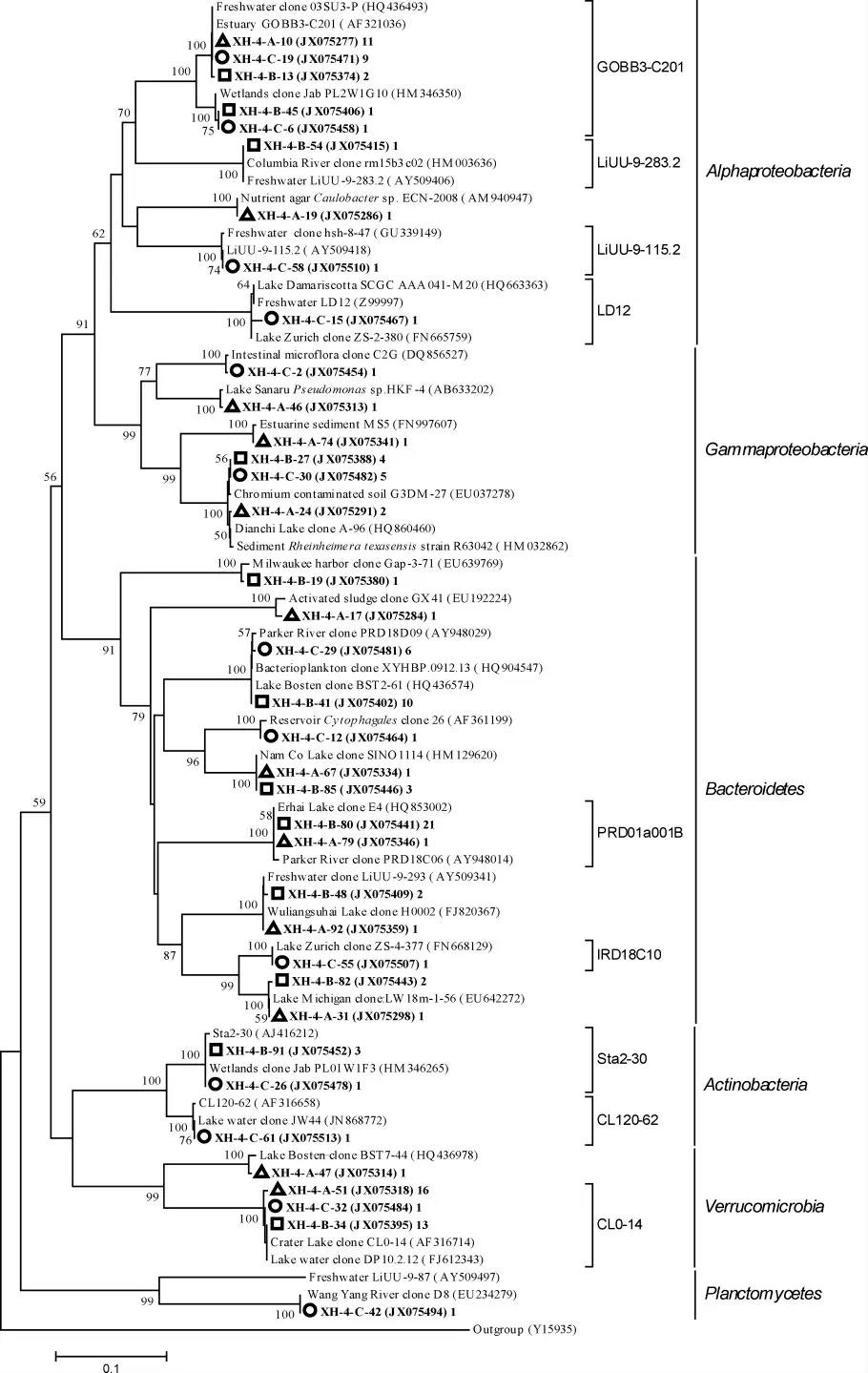
图6 除Betaproteobateria 外细菌门类基于16S rDNA 的系统发育树Fig.6 Minimum evolution trees inferred by analysis of partial 16S rDNA sequences obtained from the tree studied clone libraries shown for the other phyla except Betaproteobacteria
[1]Le C,Zha Y,Li Y et al.Eutrophication of lake waters in China:cost,causes,and control.Environment Management,2010,45(4):662-668.
[2]Smith VH,Schindler DW.Eutrophication science:where do we go from here?Ecology and Evolutionary Biology,2009,24(4):201-207.
[3]Qin B.Lake eutrophication:Control countermeasures and recycling exploitation.Ecological Engineering,2009,35(11):1569-1573.
[4]种云霄,胡洪营,钱 易.大型水生植物在水污染治理中的应用研究进展.环境污染治理技术与设备,2003,4(2):36-40.
[5]秦伯强.长江中下游浅水湖泊富营养化发生机制与控制途径初探.湖泊科学,2002,14(3):193-202.
[6]秦伯强.长江中下游湖泊富营养化发生机制与控制对策.中国科学院院刊,2007,22(6):503-505.
[7]Cotner JB,Biddanda BA.Small players,large role:microbial influence on biogeochemical processes in pelagic aquatic ecosystems.Ecosystems,2002,5(2):105-121.
[8]Ciurli A,Zuccarini P,Alpi A.Growth and nutrient absorption of two submerged aquatic macrophytes in mesocosms,for reinsertion in a eutrophicated shallow lake.Wetlands Ecology and Management,2009,17(2):107-115.
[9]Rooney N,Kalff J.Submerged macrophyte-bed effects on water-column phosphorus,chlorophyll a,and bacterial production.Ecosystems,2003,6(8):797-807.
[10]吴根福,杨志坚,周雪平等.杭州西湖水体中异养细菌生长的限制因子.应用生态学报,2003,14(8):1343-1346.
[11]董百丽,秦伯强,龚志军等.三种沉积物改良措施比较及其对苦草生长的影响.生态学杂志,2011,30(12):2726-2731.
[12]李文红,施积炎.西湖沉积物中解磷菌的分离纯化及其解磷能力.应用生态学报,2006,17(11):2112-2116.
[13]吴根福,吴 洁.杭州西湖水域微生物的生态调查.水生生物学报,2000,24(6):589-596.
[14]吴根福,虞左明,吴 洁等.杭州西湖水域可培养异养细菌的群落多样性.生物多样性,2003,11(6):467-474.
[15]Amann RI,Ludwig W,Schleifer KH.Phylogenetic identification and in situ detection of individual microbial cells without cultivation.Microbiological Reviews,1995,59(1):143-169.
[16]Muyzer G,De Waal EC,Uitterlinden AG.Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA.Applied and Environmental Microbiology,1993,59(3):695-700.
[17]吴 鑫,奚万艳,杨 虹.太湖梅梁湾冬季浮游细菌的多样性.生态学杂志,2006,25(10):1196-1200.
[18]奚万艳,吴 鑫,叶文瑾等.太湖梅梁湾水域蓝藻水华前与水华末期细菌群落结构的变化.应用与环境生物学报,2007,13(1):97-103.
[19]邢 鹏,孔繁翔,高 光.太湖浮游细菌种群基因多样性及其季节变化规律.湖泊科学,2007,19(4):373-381.
[20]Wu QL,Zwart G,Wu J et al.Submersed macrophytes play a key role in structuring bacterioplankton community composition in the large,shallow,subtropical Taihu Lake,China.Environmental Microbiology,2007,9(11):2765-2774.
[21]Tang X,Gao G,Chao J et al.Dynamics of organic-aggregate-associated bacterial communities and related environmental factors in Lake Taihu,a large eutrophic shallow lake in China.Limnology and Oceanography,2010,55(2):469-480.
[22]冯 胜,高 光,朱广伟等.基于16S rDNA-DGGE 和FDC 技术对富营养化湖泊不同生态修复工程区细菌群落结构的研究.应用与环境生物学报,2007,13(4):535-540.
[23]毛 杰,邢 鹏,刘正文.惠州西湖沉水植被修复对浮游细菌群落结构的影响.水生态学杂志,2011,32(3):26-31.
[24]金相灿,屠清瑛.湖泊富营养化调查规范:第2 版.北京:中国环境科学出版社,1990.
[25]高 光,肖 琳,杨柳燕等.环境微生物实验技术.北京:中国环境科学出版社,2004:103-107.
[26]Zhou J,Bruns MA,Tiedje JM.DNA recovery from soils of diverse composition.Applied and Environmental Microbiology,1996,62(2):316-322.
[27]Schloss PD,Westcott SL,Ryabin T et al.Introducing mothur:open-source,platform-independent,community-supported software for describing and comparing microbial communities.Applied and Environmental Microbiology,2009,75(23):7537-7541.
[28]Kim OS,Cho YJ,Lee K et al.Introducing EzTaxon-e:a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species.International Journal of Systematic and Evolutionary Microbiology,2012,62:716-721.
[29]Crump BC,Hobbie JE.Synchrony and seasonality in bacterioplankton communities of two temperate rivers.Limnology and Oceanography,2005,50(6):1718-1729.
[30]Eiler A,Bertilsson S.Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes.Environmental Microbiology,2004,6(12):1228-1243.
[31]Hahn MW.Isolation of strains belonging to the cosmopolitan Polynucleobacter necessarius cluster from freshwater habitats located in three climatic zones.Applied and Environmental Microbiology,2003,69(9):5248-5254.
[32]Zwart G,Crump BC,Agterveld MPK-V et al.Typical freshwater bacteria:an analysis of available 16S rRNA gene sequences from plankton of lakes and rivers.Aquatic Microbial Ecology,2002,28:141-155.
[33]Kasalicky'V,Jezbera J,Šimek K et al.Limnohabitans planktonicus sp.nov.and Limnohabitans parvus sp.nov.,planktonic betaproteobacteria isolated from a freshwater reservoir,and emended description of the genus Limnohabitans.International Journal of Systematic and Evolutionary Microbiology,2010,60(12):2710-2714.
[34]Vrede K.Nutrient and temperature limitation of bacterioplankton growth in temperate lakes.Microbial Ecology,2005,49(2):245-256.
[35]Morris DP,Lewis WM.Nutrient limitation of bacterioplankton growth in Lake Dillon,Colorado.Limnology and Oceanography,1992,37(6):1179-1192.
[36]Van Der Gucht K,Vandekerckhove T,Vloemans N et al.Characterization of bacterial communities in four freshwater lakes differing in nutrient load and food web structure.FEMS Microbiology Ecology,2005,53(2):205-220.
[37]冯 胜,秦伯强,高 光.细菌群落结构对水体富营养化的响应.环境科学学报,2007,27(11):1823-1829.
[38]Hitchcock JN,Mitrovic SM,Kobayashi T et al.Responses of estuarine bacterioplankton,phytoplankton and zooplankton to dissolved organic carbon (DOC)and inorganic nutrient additions.Estuaries and Coasts,2010,33(1):78-91.
[39]Van Der Gucht K,Sabbe K,De Meester L et al.Contrasting bacterioplankton community composition and seasonal dynamics in two neighbouring hypertrophic freshwater lakes.Environmental Microbiology,2001,3(11):680-690.
[40]Dirnitriu PA,Pinkart HC,Peyton BM et al.Spatial and temporal patterns in the microbial diversity of a meromictic soda lake in Washington State.Applied and Environmental Microbiology,2008,74(15):4877-4888.
[41]孙鑫鑫,刘惠荣,冯福应等.乌梁素海富营养化湖区浮游细菌多样性及系统发育分析.生物多样性,2009,17(5):490-498.
[42]Newton RJ,Jones SE,Eiler A et al.A guide to the natural history of freshwater lake bacteria.Microbiology and Molecular Biology Reviews,2011,75(1):14-49.
