基于丙型肝炎病毒非结构蛋白NS5B晶体结构的抑制剂筛选*
2013-04-18杨淑芳王中玲李真真
杨淑芳,李 爽,王中玲,李真真,傅 晟,
(1.天津经济技术开发区卫生防病站,天津 300457; 2.天津市国际生物医药联合研究院,天津 300457)
丙型肝炎病毒(hepatitis C virus, HCV)属于黄病毒科(Flaviviridae),最早发现于1989年[1],是引起慢性肝炎的主要病原体之一。目前全世界约有1.7亿HCV感染者,占全球总人口的3%[2]。其中80%感染人群会逐渐发展为慢性丙型肝炎,20%的感染者会演变成肝硬化,1%~5%的人会演变为肝细胞癌[3,4]。
丙型肝炎病毒为单股正链RNA病毒,其基因组为9.6 kb,可编码一个含有3000个氨基酸的多聚酶前体,其通过细胞和病毒的蛋白酶进行翻译后修饰等过程产生三个成熟的结构蛋白(蛋白核壳体的核心蛋白P22、包膜糖蛋白E1、E2)和六个非结构蛋白(NS2、NS3,NS4A,NS4B,NS5A,NS5B)[5]。HCV提供了许多潜在的小分子抑制靶点[6,7]。其中NS5B是HCV的RNA依赖的RNA聚合酶(RNA-dependent RNA polymerase,RdRp),是HCV复制过程中的关键酶[8]。在HCV不同的基因型和亚型中,NS5B的相关活性位点非常保守,这为筛选针对不同基因型的HCV的药物提供了重要的依据。人类细胞中不表达与NS5B RdRp功能相近的酶,使针对NS5B RdRp的抑制剂具有良好的选择性[9],因此对NS5B抑制剂的研究已经成为研究HCV感染的主要努力方向。
以NS5B为靶点的抗病毒药物分为核苷类抑制剂(nucleoside analogue inhibitor,NI)和非核苷类抑制剂( nonnucleoside analogue inhibitor,NNI)两类。如今,已有多种NI和NNI进入临床试验,如Idenix公司的IDX-184、Roche公司的R7128、Boehringer Ingelheim 公司的BI207127、Vertex公司的VCH-759等。目前为止,大部分抑制剂的开发进行到体外酶活测定阶段。本研究中筛选出的抑制剂与NS5B的复合体三维体结构将为进一步了解HCV NS5B蛋白分子抑制机制提供重要依据,也为HCV化合物药物设计提供了依据。
1 实验材料
1.1菌种与质粒 大肠杆菌(E coli)DH5α、BL21(DE3)购自TransGen Biotech公司;HCV NS5BΔ21质粒由天津市国际生物医药联合研究院高通量分子药物筛选中心提供;表达载体pET-21b质粒购自Invitrogen公司。
2 实验方法
2.1重组质粒的构建及蛋白表达纯化 以HCV NS5BΔ21基因为模板,使用Primer Premier 5.0软件设计引物,5’端引物为CCATATGTCAATGTCCTACACATGGAC,3’端引物为CCTCGAGACGAGACAGGCTGT GATATA。扩增后的PCR产物和pET-21b载体使用FastDigest Nde I和FastDigest Xho I双酶切后进行连接。
重组质粒经基因测序后转化至大肠杆菌BL 21(DE3)感受态细胞。挑取单克隆于5 ml含有100 μg/ml氨苄青霉素的LB培养基中,于37 ℃,220 r/min摇床培养8 h后按0.6%接种量接种到800 ml LB培养基中,相同条件下培养至OD 600为0.6~0.8,将摇床降温至16 ℃,加入160 μl 1 mol/L的IPTG溶液诱导后继续培养16 h,离心收集菌体(4 ℃,5500 r/min)。采用Ni亲和层析柱初步纯化蛋白,MCAC-50(20 mmol/L Tris,pH 8.0;500 mmol/L NaCl;10%甘油;50 mmol/L咪唑)洗脱杂蛋白,MCAC-500(20 mmol/L Tris pH 8.0;500 mmol/L NaCl;10%甘油;500 mmol/L咪唑)洗脱NS5B融合蛋白。通过阳离子交换层析(Resource S)进一步纯化蛋白,低盐缓冲液(300 mmol/L NaCl,20 mmol/L MES,pH 6.5)平衡系统,高盐缓冲液(700 mmol/L NaCl,20 mmol/L MES,pH 6.5)洗脱NS5B蛋白。最后将得到的NS5B蛋白溶于结晶缓冲液[600 mmol/L NaCl,10%(v/v)甘油,5 mmol/L DTT,10 mmol/L Tris,pH 7.5]中。
2.2晶体生长 现阶段用于蛋白质结晶的微量结晶技术主要有3种:气相扩散法、液液扩散法以及透析法,气相扩散法又分为坐滴法和悬滴法。本实验使用悬滴法进行晶体的生长和优化。蛋白质结晶受蛋白质自身性质和所处环境的影响,任何一个细微的变化都会影响晶体的形成及质量。本实验对NS5B蛋白浓度(25、30、35和40 mg/ml),pH值(5.0、6.0、7.0和8.0),PEG 4000(15%、20%、25%和30%)及PEG 3350(15%、20%、25%和30%),生长温度(4、16 ℃、室温)5个因素进行了梯度优化。
为了得到稳定的、质量较好、数量较多的晶体,采取Terese Bergfors提出的种晶(seeding)[10]的方法对晶体进行进一步的优化。在1.5 ml离心管中放入磁珠和200 μl晶体生长母液,用晶体环将刚长出的晶体捞出并转移至离心管中,充分震荡离心管使晶体破碎,然后用猫的胡须蘸取晶核,连续快速的滑过已经平衡好的新的液滴。
2.3基于NS5B蛋白晶体结构的抑制剂筛选
2.3.1DMSO浓度的确定本实验筛选的小分子片段库中含有384种具有苗头化合物性质(“lead-like”)的小分子化合物,都是非核苷类化合物。化合物的相对分子质量从100~200 kDa不等,平均相对分子质量为160 kDa,用DMSO溶解,浓度为50 mmol/L。DMSO是助溶剂,由于浸泡过程中加入的化合物都溶解在95%的DMSO中,高浓度的DMSO本身对NS5B蛋白晶体有损坏作用,因此在进行浸泡工作之前需要测定NS5B蛋白晶体对DMSO的耐受程度。在尽可能保证晶体质量的基础上,使DMSO浓度达到最优化,同时使浸泡时化合物的浓度最大。用95%的DMSO将晶体池液稀释至浓度分别为4.75%、2.375%、1.1875%、0.95%、0.475%和0%。然后用尼龙晶体环将生长好的NS5B蛋白晶体取出,分别放入稀释好的池液中,浸泡时间为2~24 h,每隔1 h观察1次,至晶体出现裂痕。然后进行X射线衍射数据的收集与处理。
2.3.2抑制剂筛选蛋白质与其底物、核苷酸或者小分子化合物结合后可以使其构象更加稳定,晶体的生长更加容易,近年来已有大量利用共结晶技术成功筛选到晶体的报道[11]。本实验中的晶体药物浸泡是将蛋白质分子与其底物混合在一起共同结晶的技术。该技术可以在原子水平上显示蛋白质分子与其底物复合物的结构,可以用来指导设计开发有针对性和高效性的抑制剂。
在“2.3.1”项下筛选出的合适DMSO浓度的基础上,用晶体池液将化合物稀释40倍,取2 μl 滴到玻片上,然后用尼龙晶体环将生长好的蛋白晶体从池液中捞出,加入含有化合物的池液中进行浸泡,盖上玻片。每隔1 h观察1次,直至晶体出现裂痕,然后进行X射线衍射数据的收集与处理。
3 实验结果
3.1质粒的构建及蛋白表达纯化鉴定1.7 kb的NS5B目的基因和5.4 kb的pET-21 b表达载体连接后转入大肠杆菌DH5α中,挑取阳性单克隆提取质粒后用FastDiges○RNde I和 FastDigest○RXho I进行双酶切鉴定。从图1中可以看到5.4 kb的载体条带和1.7 kb的目的基因条带,确定了重组的成功。
采用Ni亲和层析柱初步纯化蛋白,然后通过Resource S离子交换层析进一步纯化蛋白,可以得到大小为62 kDa的NS5B蛋白,蛋白的纯度可到达95%。图2显示蛋白的纯度已经达到晶体生长需要的纯度。
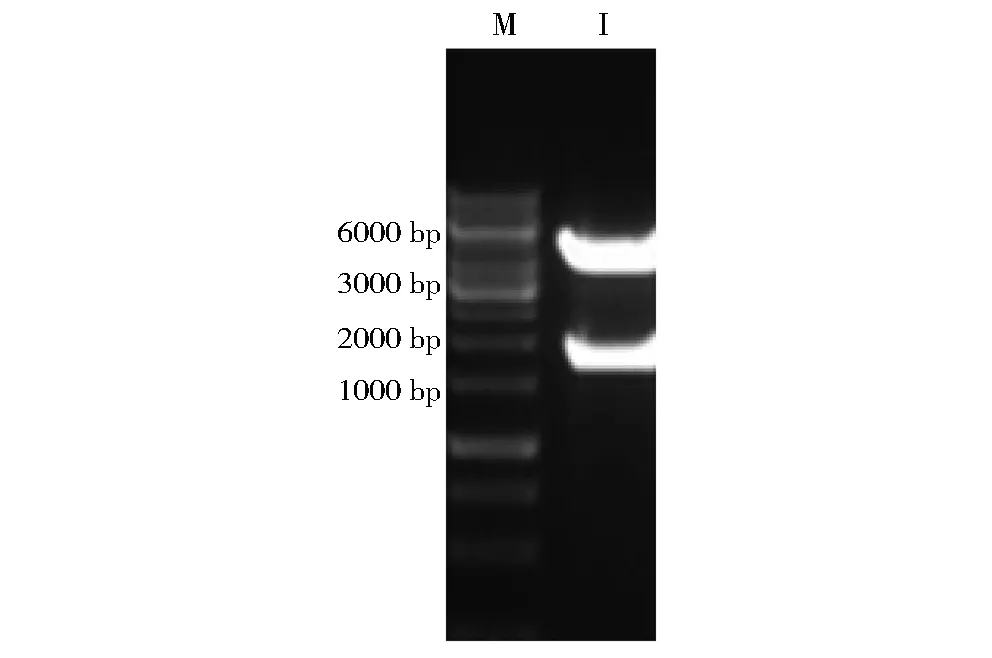
M:DNA分子量标准
3.2晶体生长结果经过对NS5B蛋白结晶条件的优化,确定了稳定单晶的生长条件:30 mg/ml NS5B蛋白,20%(w/v)PEG 4000,10%(v/v)甘油,5 mmol/L DTT,50 mmol/L MES,pH 5.5。经过种晶技术优化,得到尺寸较大(通常为3 mm×1.5 mm×1 mm),棱角分明的透明单晶,晶体的衍射分辨率达到1.9 Å。使用的防冻液为:25%乙二醇,30% PEG 4000(w/v)。采集NS5B蛋白晶体X 射线衍射数据,计算获得蛋白的三维结构。图3显示的是优化后单晶的形态。

M:蛋分子量标准 I:NS5B蛋白
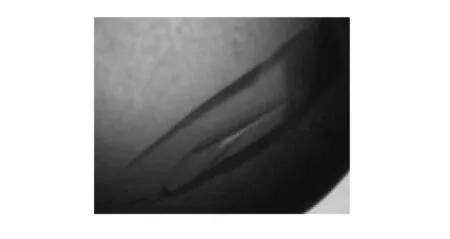
图3 NS5B单晶
3.3基于NS5B蛋白晶体结构的抑制剂筛选结果
3.3.1DMSO浓度的确定使用含有不同浓度的DMSO的池液进行晶体浸泡。从图4(A)中可以看到含有0%的DMSO晶体池液对NS5B蛋白晶体的生长没有影响;图4(B)显示晶体池液中DMSO浓度为2.375%时对NS5B蛋白晶体的生长几乎没有影响;图4(C)显示晶体池液中DMSO浓度为4.75%时,NS5B蛋白晶体明显出现了裂痕,对晶体的生长影响较大。由此确定了进行晶体浸泡时的条件:用95%DMSO溶解的化合物稀释40倍,使晶体池液中的DMSO浓度为2.375%,化合物浓度为5 mmol/L。
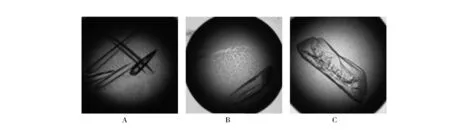
0%DMSO晶体泡药1 h2.375%DMSO晶体泡药1 h4.75%DMSO晶体泡药1 h
图4不同浓度DMSO对NS5B晶体的影响
3.3.2抑制剂筛选NS5B具有典型的“指掌”结构,主要由三部分组成,分别为拇指(thumb)区、手掌(palm)区和手指(fingers)区,其中“palm”区是核苷酸转移反应的催化中心[12]。NS5B主要有五个非核苷类抑制剂的结合位点thumb-1、thumb-2、palm-1、palm-2和palm-3[13]。构成PalmⅠ结合位点的氨基酸有Phe 193、Pro 197、Arg 200、Ser 228、Gln 291、Asn 316、Gly 317、Asp 318、Cys 366、Ser 368、Met 414、Tyr 415、Gln 446、Ile 447、Tyr 448、Gly 449 及Ser 556。其氨基酸残基Pro 197、Arg 200、Cys 366、Ser 368、Leu 384、Met 414、Tyr 415和Tyr 448,可以组成亲脂口袋,作为抑制剂结合的部位。PalmⅡ结合位点的氨基酸有Phe193、Pro 197、Arg 200、Leu 204、Leu 314、Leu 360、3Ile 63、Val 321、Ser 365、Cys 366、Val 370、Me 414 t、Ty 415r和Tyr 448。
小分子片段库中的非核苷类化合物与NS5B蛋白经过共晶后得到复合物晶体,经X射线衍射,比较收集处理的数据与NS5B蛋白母体单晶相同位置的数据,结果显示有10种小分子化合物与NS5B蛋白有结合作用。这10中小分子化合物的结构式和分子量如表1所示。其中化合物1、4、9、10与化合物的相互作用较强。
经过密度对比,化合物1结合在NS5B的Palm Ⅰ结合位点处,如图5所示。化合物1的氨基氮与NS5B的Leu384的羧基氧,吡唑环上的氮与Leu384的氨基氮分别形成氢键,结合力较强。
化合物4结合与NS5B蛋白的结合位点在Palm Ⅰ的活性口袋中,如图6所示,NS5B的Met414的甲基碳和化合物4的羰基氧形成氢键,相互作用较强。
化合物9结合在Palm Ⅱ位点,如图7所示,化合物9的氰基与NS5B蛋白的Tyr415上氧形成氢键,吗啉环上的氧原子与Cys366的硫原子形成共价键。
化合物10的六元环与NS5B的His502的咪唑环形成π-π共轭,阻碍NS5B形成二体化活性单位,从而达到抑制NS5B活性的目的。如图8所示。
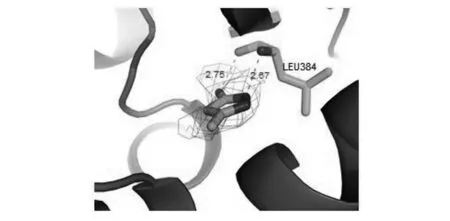
图5 化合物1与NS5B蛋白的相互作用
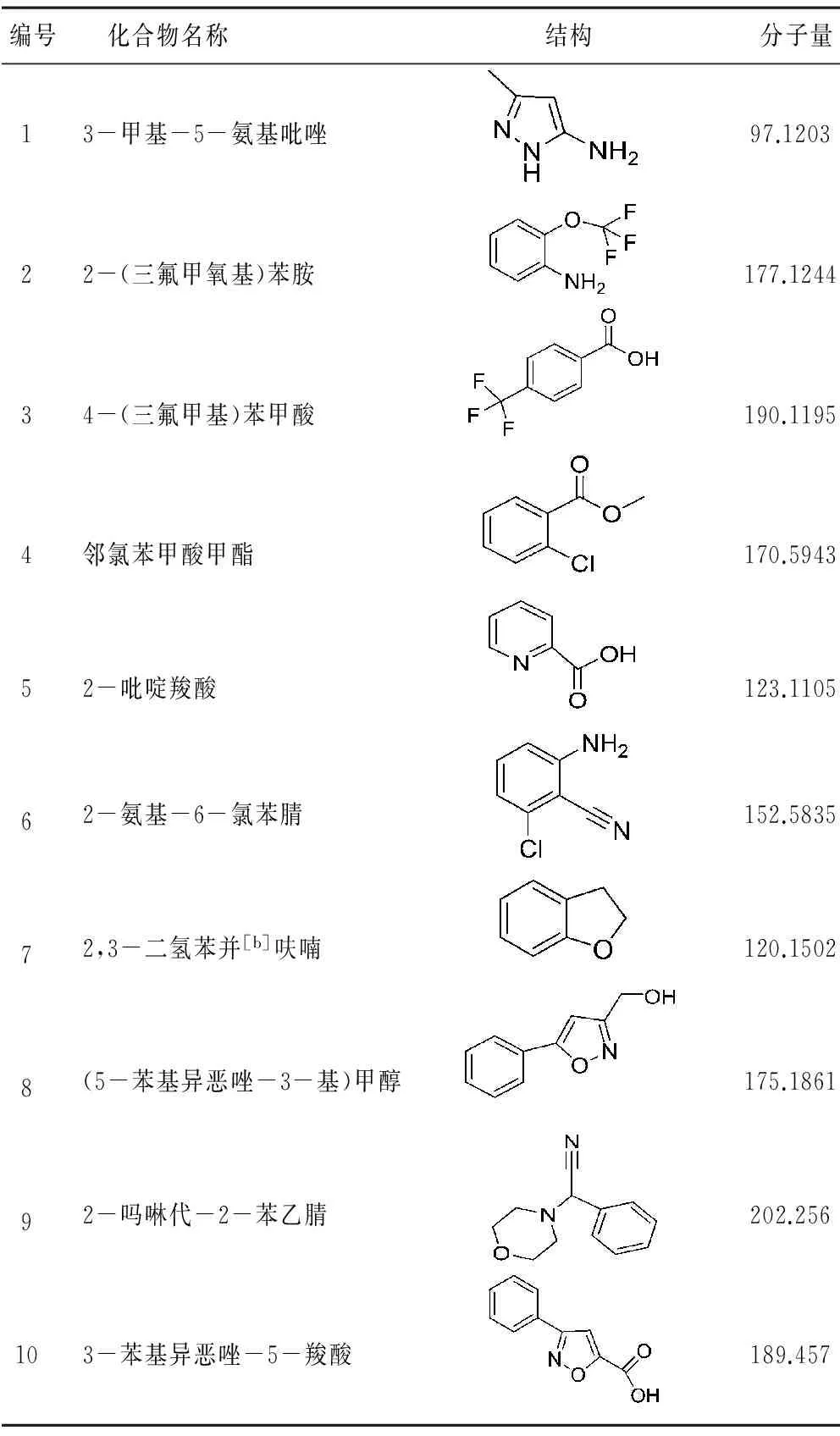
编号 化合物名称结构 分子量123456789103-甲基-5-氨基吡唑2-(三氟甲氧基)苯胺4-(三氟甲基)苯甲酸邻氯苯甲酸甲酯2-吡啶羧酸2-氨基-6-氯苯腈2,3-二氢苯并[b]呋喃(5-苯基异恶唑-3-基)甲醇2-吗啉代-2-苯乙腈3-苯基异恶唑-5-羧酸97.1203177.1244190.1195170.5943123.1105152.5835120.1502175.1861202.256189.457
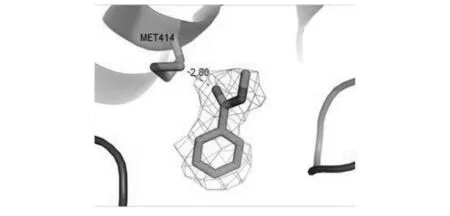
图6 化合物4与NS5B蛋白的相互作用
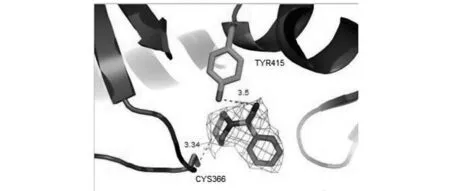
图7 化合物9与NS5B蛋白的相互作用
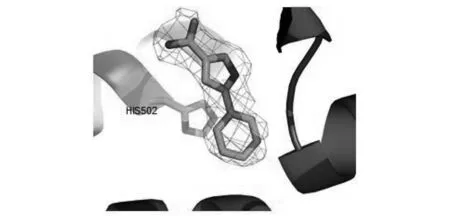
图8 化合物10与NS5B蛋白的相互作用
4 讨论
本实验将NS5B基因重组至pET-21b载体,转入大肠杆菌进行表达并得到纯度约为95%的NS5B蛋白。经过优化得到分辨率1.9 Å的NS5B晶体,在此基础上使用晶体药物浸泡的方法从小分子片段库中筛选出10种小分子抑制剂,并根据NS5B与小分子抑制剂复合物的晶体结构,揭示了化合物与靶点的结合方式和作用位点,为开发HCV药物提供了依据。
目前,在临床试验中主要使用联合用药的方法解决药物对HCV 基因型的选择性和丙型肝炎病毒对NS5B抑制剂的耐药性。一是结合于不同变构位点的没有交叉耐药性的非核苷类抑制剂联合用药;二是标准疗法聚乙二醇化α-干扰素与核苷类抑制剂或非核苷类抑制剂的联合用药;三是核苷类抑制剂与非核苷类抑制剂的联合用药。
到目前为止,临床上仍然没有有效的预防和治疗HCV的药物,采用的标准HCV治疗方案是α-干扰素(interferon alfa,,IFN-α)或聚乙二醇化α-干扰素(pegylated interferon alpha, PEG-IFNα)和利巴韦林(ribavirin, RBV)联合使用,但治疗效率低,而且存在严重不良反应。因此,迫切需要发现新型有效的抗HCV药物。近年来,寻找HCV特定靶向抗病毒治疗(specifically targeted antiviral therapy for HCV,STAT-C)药物是抗HCV研究的重要方向[14]。
已有研究表明,依赖于晶体结构的小分子片段库的筛选对分子质量低的化合物库的筛选常常能够确定新的、有吸引力苗头的化合物,这些有苗头的化合物能够进一步被优化成为潜在的具有较好成药性质的先导化合物。本实验采用的分子小片段库就是具有苗头化合物性质的化合物集合,通过晶体药物浸泡的方法筛出来的10种化合物中有4种化合物与NS5B蛋白有较强的结合作用,从原子水平上阐释了化合物与NS5B蛋白相互作用的机理,而且这些化合物可作为苗头化合物为开发新的HCV药物提供有益线索。
1Choo Q L, Kuo G, Weiner A J,etal. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome[J]. Science. 1989, 244(4902): 359
2Cheng K C, Gupta S, Wang H,etal. Current drug discovery strategies for treatment of hepatitis C virus infection[J]. J Pharm Pharmacol, 2011, 63(7): 883
3Saito I, Miyamura T, Ohbayashi Aetal. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma[J]. Proc Natl Acad Sci, 1990, 87(17): 6547
4Lauer G M, Walker B D. Hepatitis C virus infection[J]. N Engl J Med, 2001, 345(1): 41
5Hügle T, Cerny A. Current therapy and new molecular approaches to antiviral treatment and prevention of hepatitis C[J]. Rev Med Virol, 2003, 13(6): 361
6Pockros, Paul J. Developments in the treatment of chronic hepatitis C[J]. Expert Opin Investig Drugs, 2002, 11(4): 515
7Bartenschlager R, Lohmann V. Replication of the hepatitis C virus[J]. Baillieres Best Pract Res Clin Gastroenterol, 2000, 14(2): 241
8Moradpour D, Brass V, Bieck E,etal. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication[J]. J Virol, 2004, 78(23): 13278
9Beaulieu P L, Tsantrizos Y S. Inhibitors of the HCV NS5B polymerase: new hope for the treatment of hepatitis C infections[J]. Curr Opin Investig Drugs, 2004, 5(8): 838
10Bergfors T. Seeds to crystals[J]. J Struct Biol, 2003, 142(1): 66
11Erlanson D A, McDowell R S, O'Brien T. Fragment-based drug discovery[J]. J Med Chem, 2004, 47(14): 3463
12Bougie I, Charpentier S, Bisaillon M. Characterization of the metal ion binding properties of the hepatitis C virus RNA polymerase[J]. J Biol Chem, 2003, 278(6): 3868
13Pessoa M G, Wright T L. Update on clinical trials in the treatment of hepatitis B[J]. J Gastroenterol Hepatol, 1999, 14(5s): 6
14Lange C M, Sarrazin C, Zeuzem S. Review article: specifically targeted anti-viral therapy for hepatitis C - a new era in therapy[J]. Aliment Pharmacol Ther, 2010, 32(1): 14
