毛壳素显著抑制柯萨奇病毒B3的体外复制
2013-01-14张世仑张媛媛尹隽钟江
张世仑,张媛媛,尹隽,钟江
复旦大学生命科学学院微生物学与微生物工程系,上海 200433
毛壳素(chaetocin)是最早分离自毛壳属(Chaetomium)真菌的一种天然代谢产物[1],属表硫代二酮吡嗪复合物(epidithiodiketopiperazine),因其具有特殊的生物活性而受到重视。
研究表明,毛壳素是真核细胞组蛋白甲基转移酶Su(VAR)3-9和G9a的特异性抑制剂[2,3],可改变细胞染色质结构,影响细胞的表观遗传学调控[4],从而改变细胞一些重要基因的表达。在一些肿瘤细胞中,毛壳素可影响若干与肿瘤生长密切相关基因的表达,因此在肿瘤治疗方面有潜在临床价值[5-7]。同时,毛壳素也可激活整合在染色体上的人类免疫缺陷病毒(human immunodeficiency virus,HIV)的原病毒,在清除艾滋病患者体内HIV的治疗中也可能发挥重要作用[8]。此外,有研究表明,毛壳素可抑制细胞硫氧还蛋白还原酶的活性,从而激活细胞氧化应激通路,影响细胞的生理状态和功能[9]。目前,除影响HIV的原病毒基因表达和活化外,有关毛壳素对病毒感染和复制的影响研究还未见报道。
人柯萨奇病毒B3(coxsackievirus B3,CVB3)属细小RNA病毒科(Picornaviridae)肠道病毒属(Enterovirus)B组柯萨奇病毒。其基因组是长约7.4 kb的单链RNA。许多研究表明,该病毒与人类病毒性心肌炎有密切关系[10]。目前对该病毒感染尚缺少特效药物和疫苗,故研发控制病毒感染和致病的新手段至关重要。
本文研究了毛壳素对CVB3在体外培养细胞中复制的影响,显示其可显著降低病毒的复制水平,减少细胞死亡。
1 材料与方法
1.1 病毒和细胞
CVB3毒株由本实验室保存,在HeLa细胞中扩增。HeLa细胞用含10%胎牛血清和100 U/ml青霉素、100 mg/ml链霉素的 RPMI 1640 培养液培养。Vero细胞用含同样血清和抗生素的高糖DMEM培养液培养,培养条件为37 ℃、5% CO2。
1.2 病毒感染细胞
以适量细胞接种6孔板或24孔板(6孔板,2×105个细胞/孔;24孔板,5×104个细胞/孔),使细胞覆盖度达70%左右。细胞培养过夜后,在培养液中加入一定量病毒。除特别指出外,每孔加入效价为2×106半数组织培养感染剂量(50% tissue culture infective dose,TCID50)/ml的病毒液20 μ l(6孔板)或5 μ l(24孔板),使病毒感染剂量为0.2 TCID50/细胞。采用其他剂量时也依此计算病毒液的用量。
1.3 药物处理细胞
毛壳素购自Cayman公司(美国),用二甲亚砜(dimethyl sulfoxide,DMSO)配制成浓度为1 mmol/L的溶液,处理时与病毒同时加入细胞培养液。N-乙酰半胱氨酸(N-acetyl cysteine,NAC)购自MP Biochemicals公司,配制成100 mmol/L的无菌水溶液。处理时提前加入细胞培养液,1 h 后再加入病毒及毛壳素。
1.4 病毒产量及病毒液效价测定
病毒感染HeLa细胞48 h后,将感染细胞置于-80 ℃及37 ℃下冰冻-融解3个循环。取上清液,系列稀释后接种于长有HeLa细胞的96孔培养板中,在37 ℃、5% CO2条件下培养2~3 d后,显微镜下观察细胞病理变化,按Reed-Muench法计算病毒效价(TCID50/ml)。
1.5 细胞活性分析
采用CCK8细胞活性试剂(同仁化学研究所)检测病毒感染对细胞的杀伤作用,操作按说明书进行。细胞感染24 h后,在细胞培养液中加入适量CCK8试剂,继续培养2 h,用SpectraMax M5多功能读板仪(Molecular Devices, USA)分别读取OD450 nm和OD600 nm,并取OD450 nm与OD600 nm的差值为最终读数,反映细胞的相对活性。
1.6 定量反转录聚合酶链反应
将病毒感染HeLa细胞,24 h后用RNAiso Plus(TaKaRa)总RNA提取试剂盒提取细胞总RNA,并用PrimeScript RT试剂盒(TaKaRa) 获得总cDNA,用SYBR Green实时定量聚合酶链反应(polymerase chain reaction,PCR)进行病毒cDNA 的定量分析。所用引物根据CVB3编码VP4的基因设计,具体序列为:5′ -CAAATAC-AAAGGGGCTCAAGTATCA-3′ 和5′ -GCGTCT-AGATAAGTTGAGAGCTGGTAG-3′ 。以β -actin基因作为内参,引物序列为5′ -TGGAATCCTGT-GGCATCCATGAAAC-3′ 和 5′ -TAAAACGCA-GCTCAGTAACAGTCCG-3′ 。SYBR Green试剂购自Bio-Rad公司,实时荧光定量 PCR 在Stratagene Mx Real Time PCR 仪上进行。
1.7 统计分析
所有实验均经3次以上重复验证。数据以mean±SD表示,行t检验,P<0.05为有显著性差异。
2 结果
2.1 毛壳素显著减少病毒引起的细胞死亡
在将HeLa细胞用不同剂量CVB3感染的同时,加入250 nmol/L毛壳素,并将不加毛壳素的感染细胞设为对照。较高剂量感染时,24 h后检测病毒感染导致细胞死亡的情况;低剂量感染时,48 h后检测相应细胞死亡的情况。结果如图1所示。病毒感染造成细胞严重死亡,与对照组相比,加入毛壳素明显抑制病毒感染引起的细胞死亡,提高细胞相对存活率(P<0.01)。当感染剂量为0.5 TCID50/细胞时,24 h后细胞相对存活率从未加毛壳素的(21.9±1.8)%提高至(70.1±4.3)%。采用其他感染剂量时,毛壳素均有类似的抑制病毒引起细胞死亡的效果。

MOI, multiplicity of infection (TCID50/cell). A: 24 h post-infection. B: 48 h post-infection.
图1毛壳素(250 nmol/L)对不同剂量CVB3感染的HeLa细胞相对存活率的影响(n=6)
Fig.1 Effect of chaetocin treatment (250 nmol/L) on the relative cell viability of HeLa cells infected with different dosages of CBV3 (n=6)
进一步用感染剂量为0.5 TCID50/细胞的CVB3感染HeLa细胞,同时加入不同剂量的毛壳素,24 h后检测病毒感染造成的细胞死亡情况。结果如图2所示。当毛壳素浓度在500 nmol/L及以下时,未对HeLa细胞生长造成明显影响。未加毛壳素时,病毒感染导致细胞相对存活率降低至对照组的(41.1±0.8)%;随着毛壳素浓度上升,病毒感染细胞的相对存活率上升,至毛壳素浓度为250 nmol/L时,细胞相对存活率上升至(80.9±3.8)%;但随着毛壳素浓度进一步上升,细胞的相对存活率不再提高,且由于高浓度药物的细胞毒性,细胞相对存活率甚至降低(未显示)。以上结果表明,毛壳素可抑制病毒感染造成的细胞死亡。

图2不同浓度毛壳素对CVB3感染的HeLa细胞相对存活率的影响(n=3)
Fig.2 Effect of different concentrations of chaetocin on the relative cell viability of HeLa cells infected with CBV3 (n=3)
为了解毛壳素的作用是否仅限于HeLa细胞,本研究还采用CVB3感染非洲绿猴肾细胞(Vero细胞),同时加入200 nmol/L毛壳素。以未感染也未加毛壳素的细胞作为参照(细胞存活率设为100%),加入毛壳素但未感染的细胞相对存活率为(91.0±6.4)%,表明药物对Vero细胞的活力有一定影响,但仍有限。在病毒感染的同时加入毛壳素,细胞的相对存活率为(87.2±4.8)%,显著高于未加入毛壳素的感染细胞的(64.6±1.7)%(P<0.01)。结果表明,毛壳素对CVB3感染Vero细胞也有一定的抑制作用。
2.2 毛壳素显著抑制子代病毒的产量
在病毒感染细胞的同时,加入100、250 nmol/L毛壳素,以DMSO组为对照,48 h后测定病毒效价。结果如图3所示。不同浓度的毛壳素均降低病毒产量(250 nmol/L时P<0.01)。毛壳素浓度为250 nmol/L时,病毒产量仅为对照组的(5.3±0.8)%。
2.3 毛壳素显著抑制病毒RNA复制水平
在加入和不加入毛壳素的情况下,分别用CVB3感染HeLa细胞,48 h 后提取感染细胞的总RNA,用实时定量PCR检测细胞中病毒RNA的相对水平。结果如图4所示。毛壳素使病毒RNA复制水平降低至不加时的(13.0±8.3)%,显著抑制病毒RNA复制(P<0.05)。

图3毛壳素和N-乙酰半胱氨酸对CVB3感染HeLa细胞中CVB3子代病毒产量的影响(n=3)
Fig.3 Effect of chaetocin andN-acetyl cystine on progeny virus production in CVB3-infected HeLa cells (n=3)

图4毛壳素对CVB3感染HeLa细胞中病毒RNA复制的影响(n=4)
Fig.4 Effect of chaetocin on CVB3 RNA replication in CVB3-infected HeLa cells (n=4)
2.4 NAC对毛壳素抑制病毒复制作用的影响
鉴于有研究提示毛壳素可能通过激活细胞的氧化应激反应来影响细胞状态,从而提高细胞抵御病毒感染的能力,本研究在加入毛壳素的同时加入抑制氧化应激通路的还原剂NAC,分析氧化应激通路的作用。结果如图3和图5所示。1 mmol/L NAC对CVB3 感染造成的细胞死亡没有影响(图5),在加入毛壳素的同时加入NAC可部分减弱毛壳素的作用,细胞相对存活率有所降低(P<0.01,图5),病毒产量相应提高(P<0.01,图3),但细胞相对存活率仍与未加毛壳素时有显著差异(P<0.01, 图5)。
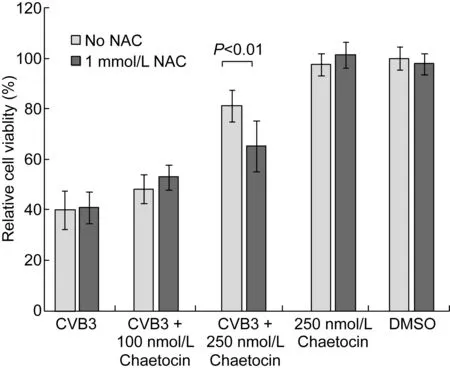
图5N-乙酰半胱氨酸对毛壳素抑制CVB3复制的影响(n=3)
Fig.5 Effect ofN-acetyl cystine on the inhibition of CVB3 replication by chaetocin (n=3)
3 讨论
本研究比较了病毒引起的细胞死亡、子代病毒产量及细胞内病毒RNA拷贝数。结果显示,毛壳素可显著抑制CVB3在HeLa细胞中的RNA复制、子代病毒产量及细胞死亡。250 nmol/L毛壳素可使CVB3感染的HeLa细胞相对存活率从(21.9±1.8)%提高至(70.1±4.3)%,病毒产量降低至对照组的(5.3±0.8)%,而病毒RNA水平也仅为对照组的(13.0±8.3)%。毛壳素对CVB3在Vero细胞中的复制也有一定抑制作用,使CVB3感染的Vero细胞相对存活率从(64.6±1.7)%提高至(87.2±4.8)%,提示该作用具有普遍性。尽管如此,毛壳素在其他细胞乃至动物模型中的作用仍有待深入研究。
本研究结果表明,在细胞培养系统中,低浓度毛壳素(低于250 nmol/L或174 ng/ml)就能有效抑制病毒复制。在使用的浓度范围内,毛壳素对HeLa细胞活力影响甚微(图2);200 nmol/L毛壳素对Vero细胞的作用也有限,细胞存活率仅降至未处理时的(91.0±6.4)%。据文献报道,毛壳素在小鼠中经口急性半数致死剂量为1 200 mg/kg体重。因此,本实验所使用的浓度在安全范围内。
毛壳素在细胞中的生物活性有2个方面:①抑制组蛋白甲基转移酶的活性,降低组蛋白H3K9的甲基化水平,从而改变细胞的表观遗传学调控,影响宿主细胞基因表达。②抑制细胞硫氧还蛋白还原酶的活性,改变细胞的氧化应激状态。
表观遗传学调控机制是细胞基因表达调控的重要机制。已有研究表明,许多DNA病毒的复制受细胞表观遗传机制的影响[11,12]。如乙型肝炎病毒复制中形成的共价闭合环状DNA(covalently closed circular DNA,cccDNA)在细胞核中与细胞组蛋白结合形成核小体[13],且组蛋白的修饰状态对乙型肝炎病毒基因转录和复制有相当大的影响[14,15]。腺相关病毒(adeno-associated virus)载体 在细胞中会形成游离子染色质(episomal chromatin)[16]。DNA甲基化和组蛋白乙酰化也会影响EB病毒(Epstein-Barr virus,EBV)启动子的活性,导致病毒在隐性感染与裂解性感染之间发生转变[17,18]。
对RNA病毒的感染和复制是否受表观遗传学机制的影响很少有研究。事实上,由于表观遗传学机制会影响细胞基因的表达,因此有可能间接影响病毒的复制和致病性。最新报道发现,组蛋白去乙酰化酶抑制剂——伏立诺他(suberoylanilide hydroxamic acid,SAHA)可通过上调骨桥蛋白(osteopontin)和下调载脂蛋白A1 (apolipoprotein A1)的水平来抑制丙型肝炎病毒在体外细胞系中的复制,表明调节细胞表观遗传学机制有可能成为治疗RNA病毒感染的新途径[19]。
另一方面,细胞的氧化还原状态在细胞信号转导、基因表达调控等方面发挥重要作用,也与病毒复制密切相关。病毒感染往往伴随细胞氧化状态变化[20-22],这种变化既可以是病毒感染诱导的,也可以是细胞为对抗病毒感染所作出的反应。有研究表明,由于缺少重要的微量元素锶引起的氧化应激,导致柯萨奇病毒的危害更严重[23]。也有实验表明,加入抗氧化剂NAC可一定程度抑制肠道病毒71(enterovirus 71,EV71)在Vero 细胞中的复制和病毒产量[24]。本研究结果表明,在毛壳素处理的同时加入NAC,可一定程度抵消毛壳素抑制CVB3复制和提高细胞存活率的作用(图2、5),但并未完全消除毛壳素的作用,具体作用机制有待深入探讨。
总之,本研究结果提示,毛壳素可明显降低CVB3在HeLa细胞中的复制水平,并减少CVB3对细胞的损伤。对其抑制病毒机制的深入研究将有助于更好地认识病毒的复制机制,以及病毒与细胞在表观遗传学和氧化应激方面的相互作用;同时也有可能为研发新的抗病毒药物提供线索。
[1] Udagawa S, Muroi T, Kurata H, Sekita S, Yoshihira K, Natori S, Umeda M. The production of chaetoglobosins, sterigmatocystin, O-methylsterigmatocystin, and chaetocin by Chaetomium spp. and related fungi [J]. Can J Microbiol, 1979, 25(2):170-177.
[2] Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 [J]. Nat Chem Biol, 2005, 1(3):143-145.
[3] Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity [J]. J Am Chem Soc, 2010, 132(12):4078-4079.
[4] Illner D, Zinner R, Handtke V, Rouquette J, Strickfaden H, Lanctt C, Conrad M, Seiler A, Imhof A, Cremer T, Cremer M. Remodeling of nuclear architecture by the thiodioxoxpiperazine metabolite chaetocin [J]. Exp Cell Res, 2010, 316(10):1662-1680.
[5] Chaib H, Nebbioso A, Prebet T, Castellano R, Garbit S, Restouin A, Vey N, Altucci L, Collette Y. Anti-leukemia activity of chaetocin via death receptor-dependent apoptosis and dual modulation of the histone methyl-transferase SUV39H1 [J]. Leukemia, 2012, 26(4):662-674.
[6] Lee YM, Lim JH, Yoon H, Chun YS, Park JW. Antihepatoma activity of chaetocin due to deregulated splicing of hypoxia-inducible factor 1α pre-mRNA in mice and in vitro [J]. Hepatology, 2011, 53(1):171-180.
[7] Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress [J]. Blood, 2007, 109(6):2579-2588.
[8] Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response [J]. FEBS Lett, 2011, 585(22):3549-3554.
[9] Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC. The anticancer agent chaetocin is a competitive substrate and inhibitor of thioredoxin reductase [J]. Antioxid Redox Signal, 2009, 11(5): 1097-1106.
[10] Rose NR. Myocarditis: infection versus autoimmunity [J]. J Clin Immunol, 2009, 29(6):730-737.
[11] Lieberman PM. Chromatin regulation of virus infection [J]. Trends Microbiol, 2006, 14(3): 132-140.
[12] Lieberman PM. Chromatin organization and virus gene expression [J]. J Cell Physiol, 2008, 216(2):295-302.
[13] Bock CT, Schranz P, Schröder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell [J]. Virus Genes, 1994, 8(3):215-229.
[14] Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G, Levrero M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones [J]. Gastroenterology, 2006, 130(3):823-837.
[15] Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection [J]. J Hepatol, 2009, 51(3):581-592.
[16] Penaud-Budloo M, Le Guiner C, Nowrouzi A, Toromanoff A, Chérel Y, Chenuaud P, Schmidt M, von Kalle C, Rolling F, Moullier P, Snyder RO. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle [J]. J Virol, 2008, 82(16):7875-7885.
[17] Takacs M, Banati F, Koroknai A, Segesdi J, Salamon D, Wolf H, Niller HH, Minarovits J. Epigenetic regulation of latent Epstein-Barr virus promoters [J]. Biochim Biophys Acta, 2010, 1799(3-4):228-235.
[18] Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome [J]. Proc Natl Aacd Sci USA, 2010, 107(2): 850-855.
[19] Sato A, Saito Y, Sugiyama K, Sakasegawa N, Muramatsu T, Fukuda S, Yoneya M, Kimura M, Ebinuma H, Hibi T, Saito H. Suppressive effect of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), on hepatitis C virus replication via epigenetic changes in host cells [J]. J Cell Biochem, 2013 [Epub ahead of print].doi: 10.1002/jcb.24541.
[20] Schwarz KB. Oxidative stress during viral infection: a review [J]. Free Radic Biol Med, 1996, 21(5):641-649.
[21] Nencioni L, Sgarbanti R, De Chiara G, Garaci E, Palamara AT. Influenza virus and redox mediated cell signaling: a complex network of virus/host interaction [J]. New Microbiol, 2007, 30(4):367-375.
[22] Ivanov AV, Bartosch B, Smirnova OA, Isaguliants MG, Kochetkov SN. HCV and oxidative stress in the liver [J]. Viruses, 2013, 5(2):439-469.
[23] Beck MA. Selenium and host defence towards viruses [J]. Proc Nutr Soc, 1999, 58(3):707-711.
[24] Ho HY, Cheng ML, Weng SF, Chang L, Yeh TT, Shih SR, Chiu DT. Glucose-6-phosphate dehydrogenase deficiency enhances enterovirus 71 infection [J]. J Gen Virol, 2008, 89 (Pt 9):2080-2089.
