Complete mitogenome of the Lesser Purple Emperor Apatura ilia (Lepidoptera: Nymphalidae: Apaturinae) and comparison with other nymphalid butterflies
2012-12-25CHENMeiTIANLiLiSHIQingHuiCAOTianWenHAOJiaSheng
CHEN Mei, TIAN Li-Li, SHI Qing-Hui, CAO Tian-Wen, HAO Jia-Sheng,*
(1. College of life Sciences, Anhui Normal University, Wuhu Anhui 241000, China; 2. Institute of Plant Protection, Shanxi Academy of Agriculture Science, Taiyuan Shanxi 030031, China)
Complete mitogenome of the Lesser Purple EmperorApatura ilia(Lepidoptera: Nymphalidae: Apaturinae) and comparison with other nymphalid butterflies
CHEN Mei1, TIAN Li-Li1, SHI Qing-Hui1, CAO Tian-Wen2,*, HAO Jia-Sheng1,*
(1.College of life Sciences,Anhui Normal University,Wuhu Anhui241000,China; 2.Institute of Plant Protection,Shanxi Academy of Agriculture Science,Taiyuan Shanxi030031,China)
The complete mitochondrial genome ofApatura ilia(GenBank accession no. JF437925) was determined as a circular DNA molecule of 15 242 bp, with common genes of 13 putative proteins, 2 rRNAs, and 22 tRNAs and of the same gene arrangement as in other sequenced lepidopterans. All protein-coding genes had the typical start codon ATN, except for the COI’s using CGA as its start codon as previously demonstrated in other lepidopteran species. The comparison of the nucleotide sequences of theA. iliamitogenome with ten other Nymphalidae species showed nearly identical gene orientation and arrangement, with only a few alterations in non-coding fragments. The nucleotide composition and codon frequency all fell into the range estimated for the order Lepidoptera. TheA. iliamitochondrial genome had the canonical set of 22 tRNA genes folded in the typical cloverleaf structure, with an unique exception of tRNASer(AGN). The mitochondrial genes fromA. iliawere overlapped in a total of 33 bp at 9 locations, as well as interleaved with a total of 155 bp intergenic spacers, spread over 12 regions with the size ranging from 1 to 49 bp. Furthermore, the spacer between ND6 and Cytbharbored a microsatellite-like repeat (TA)23not found in other completely sequenced nymphalid genomes. The 403 bp AT-rich region harbored two conserved motifs (ATAGA, ATTTA), a 21 bp polyT stretch, a 10 bp poly-A region, along with two microsatellite-like repeats ( (TA)10and (TA)7), as detected in other nymphalid butterflies.
Mitochondrial genome; Lepidoptera; Nymphalidae;Apatura ilia
The mitochondrial genome of insects is a circular and double-stranded molecule of approximately 14-20 kb in size with highly conserved exon arrangement covering a set of 37 genes, namely 13 PCGs, 22 tRNA genes, and 2 rRNA (srRNA (12S) and lrRNA (16S)) genes (Boore, 1999; Taanman, 1999). A mitogenome features smaller sizes, faster evolutionary rates, higher conservative gene content, maternal inheritance and little recombination (Brown, 1983; Avise, 1994), compared to the nucleic genome. Thus it has been commonly used for taxonomic and phylogenetic studies in many animal groups. The availability of complete mitogenome data of more species remarkably increases the accuracy and efficiency of a variety of research areas, such as molecular phylogenetics, phylogeography and taxonomy.
The complete mitochondrial genomes of nearly 240 insect species are available, of which only 10 are of Papilionoidae, despite its high biodiversity of 17 500 species (Robbins, 1982). The Nymphalidae is the largest butterfly family, and relationships with other butterfly groups remain unclear. It is necessary to integrate mitogenome data in the reconstruction of Nymphalidae phylogenies. In this study we sequenced the entire mitogenome of the nymphalidA. ilia, a representative butterfly species of the subfamily Apaturinae and compared its nucleotide organization to those of other representative nymphalid butterfly species. Our aim is to provide important molecular data to clarify the phylogenetic relationship betweenA. iliaand other nymphalid butterflies.
1 Materials and Methods
1.1 Sample collection and DNA extraction
Adult individuals ofA. iliawere collected from Mount Yandangshan, Zhejiang, China in August 2008. Samples were quickly preserved in 100% ethanol and at -20 °C until DNA extraction. Total genomic DNA was isolated from a single frozen butterfly using the proteinase-K-SiO2as follows (Hao et al, 2005). The thorax muscle around 5 mm3was removed into a 10 mL Eppendorf tube, washed twice with ddH2O, and soaked for 2~3 h. Incubation was done with 500 μL DNA solution (5 mmol/L NaCl, 0.5% SDS, 15 mmol/L EDTA, 10 mmol/L Tris-HCl, pH 7.6) and 40 μL proteinase-K (20 mg/mL). The muscle was then bathed at 55 °C for 10~12 h and centrifuged at 4 000 rpm for 2 min. Liquid supernatant was transferred to a new 10 mL Eppendorf tube with 500 μL 8 mol/L GuSCN and 40 μL 50% clean glass liquid mixture, bathed at 37 °C for 1~2 h, shocked every ten min and centrifuged at 4 000 rpm for 1 min. The supernatant was removed and sediments were cleaned twice with 75% alcohol, and once with acetone. The sample was dried thoroughly in a vacuum dryer at 45 °C prior to the addition of 60 μL TE (10 mmol/L Tris-Cl, 1mmol/L EDTA, pH 8.0). The solution was later bathed at 56 °C for 30 min, and centrifuged at increasing speed untill 4 000 rpm for 1 min. The supernatant containing total genomic DNA was removed into a clean 1.5 mL Eppendorf tube and preserved at -20 °C for use.
1.2 Primer design, PCR amplification and DNA sequencing
The universal PCR primers for short fragment amplifications of the srRNA, COI and Cyt b genes were synthesized after Simon et al (1994) and Simons & Weller (2001). Long primers and certain short ones for some genes including COIII and ND5 were designed by the multiple sequence alignments of the complete mitochondrial genomes of all lepidopterans available (Tab. 1), using ClustalX 1.8 (Thompson et al, 1997) and Primer Premier 5.0 (Singh et al, 1998) softwares.
Long PCRs were performed using TaKaRa LA Taq polymerase with the cycling parameters: initial denaturation for 5 min at 95 °C, followed by 30 cycles of 95 °C for 50 sec, 47-61 °C for 50 sec, 68 °C for 2 min and 30 sec; and a final extension step of 68 °C for 10 min. The short fragments were amplified with TaKaRa Taq polymerase: initial denaturation for 5 min at 94 °C, followed by 35 cycles of 94 °C for 1 min, 45-53 °C for 1 min, 72 °C for 2 min; and a final extension step of 72 ° for 10 min. The PCR products were detected via electrophoresis in 1.2% agarose gel, purified using the 3S Spin PCR Product Purification Kit and sequenced directly with ABI–3730 automatic DNA sequencer. Mitogenome sequence data have been deposited into GenBank under the accession number JF437925.
1.3 Sequence analysis
All genes and the AT-rich region of theA. iliamitogenome ClustalX 1.8. The nucleotide sequences of protein-coding genes were translated according to the invertebrate mtDNA genetic code. Fifteen of the 22 tRNA genes were identified using the software tRNA Scan-SE 1.21 (Lowe & Eddy, 1997) and RNAstructure 4.3 (Mathews, 2006). The remaining 7 tRNA genes were drawn manually after comparison with known homologous regions of other lepidopteran insects. MEGA 5.0 software (Tamura et al, 2007) was used to analyze nucleotide composition and codon usage.
2 Results
2.1 Gennome organization
The complete mitogenome ofA. iliais 15 242 bp in size. Similar to most insects, the mitogenome has a set of 37 genes: including 13 protein-coding, 22 tRNA and 2 rRNA genes. A large noncoding A+T-rich region was identified (Fig. 1). This region is of highly variable length in insects and is generally suggested to be the replication and transcription origin sites of the mtDNA double strands (Clayton, 1992). Furthermore the mitogenome ofA. iliawas found to be highly similar to most sequenced lepidopterans in terms of gene order and orientation. Nine protein-coding genes were found in the major strand and the remaining 4 protein-coding genes in the minor strand along the mitogenome(Tab. 1). Besides those, the mitogenome ofA. iliahas 9 overlapped sequences and 12 intergenic sequences.
2.1 试题紧扣教材且高于教材 生物学教材是生物学教学的重要工具和生物学课程实施的载体,也是试题的主要来源。试题源于教材是让学生重视教材内容,要求读懂、理解和解释教材中的生物学基本概念、原理和规律等方面的基础知识。试题高于教材是对教材内容适度转化、加工提升,在试题设计的关键处突出重要概念,考查学生对知识的理解和适度迁移能力,体现用教材教的基本思想。
2.2 Protein-coding genes, transfer RNA genes and ribosomal RNA genes
Thirteen protein-coding genes for 3 711 amino acids were identified in the mitochondrial genome ofA. ilia(Tab. 2). The longest one is the COI gene with 1 533 bp and the shortest one is ATP8 with only 159 bp. Twelve protein-coding genes were initiated by conventional start codon ATN, while only the COI gene was tentatively designated to be CGA as the start codon. InA. ilia, nine protein-coding genes ended with TANs (7 with TAA, 2 with TAG), while four genes ended with a single T right ahead of tRNA genes (Tab. 1).
Twenty-two tRNA genes were found in the mitogenome (Fig. 2), all of the cloverleaf secondary structure except for tRNAser(AGN) which harbors a simple loop in the DHU arm. Twenty-two tRNA genes ranged from 62 bp for tRNAArgand tRNASer(AGN) to 71 bp for tRNALysin length(Tab. 3).
As in other lepidopteran species, theA. iliamitogenome was found to harbor two rRNA genes, srRNA (776 bp) and lrRNA (1,333 bp). They are located between tRNALeu(CUA) and an A+T-rich region, separated by tRNAVal.
2.3 A+T-rich region, intergenic spacer and overlapping sequences
The AT-rich region ofA. iliawas found to be 403 bp in length, located between srRNA and tRNAMet. It had the highest AT content (92.5%) across the whole mitogenome (Tab. 2), typical in Nymphalidae insects from 89.6% (Melnitis leda) (Unpublished) to 96.3% (Libythea celtis) (Unpublished)..In addition, the values of the AT skew and GC skew for this region reached to -0.07 and -0.14, respectively.
Thirteen intergenic spacer sequences were determined with a total length of 155 bp (Tab. 4). The longest two intergenic spacer sequences were both 49 bp long and located between tRNAGlnand ND2 and ND6 and Cytb. The shortest one was only 1 bp in size. The
mitogenome ofA. iliacontains nine overlapping sequences ranging from 1 bp to 8 bp and totaling 33 bp in length.
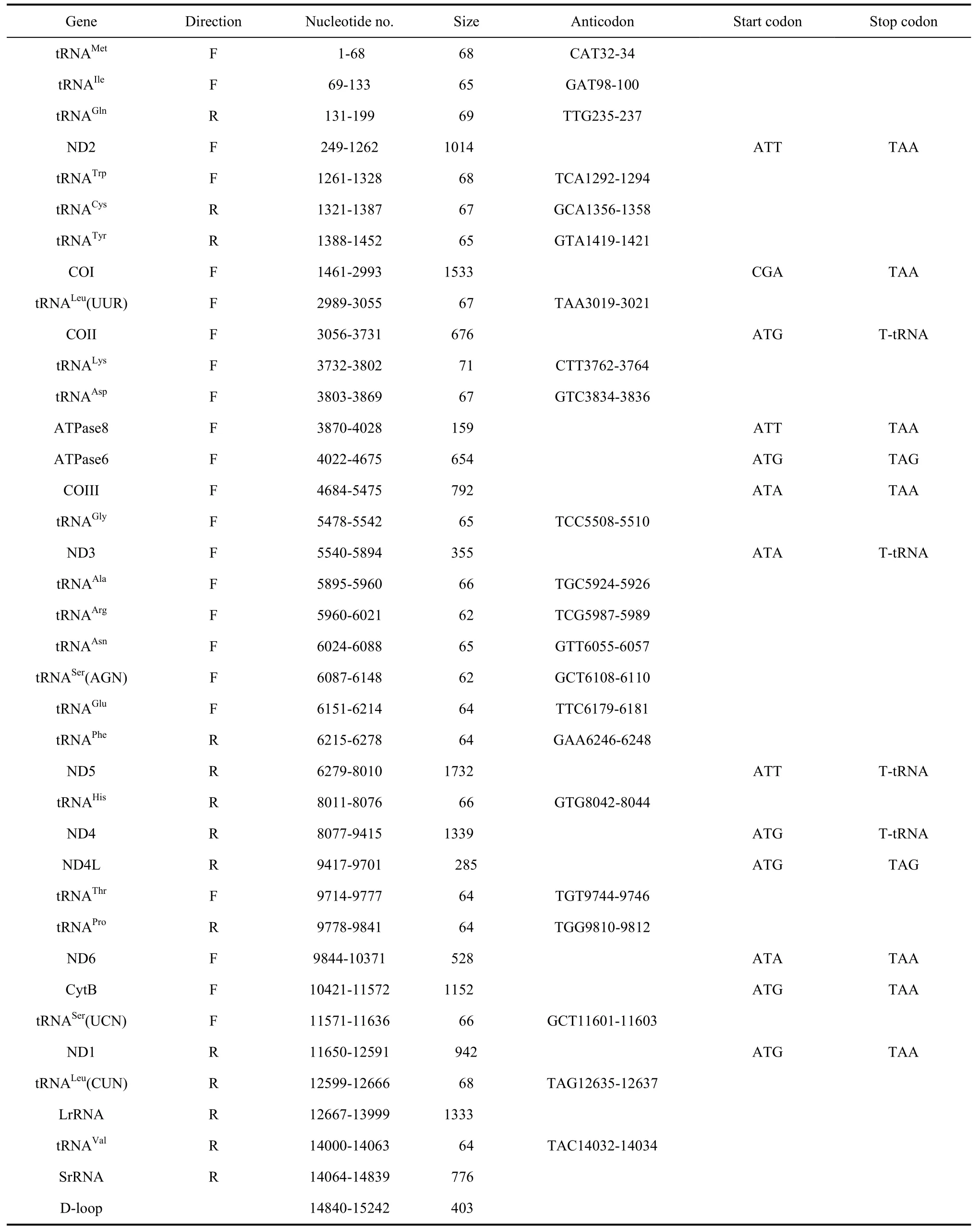
Tab. 1 Summary of the Apatura ilia mitogenome
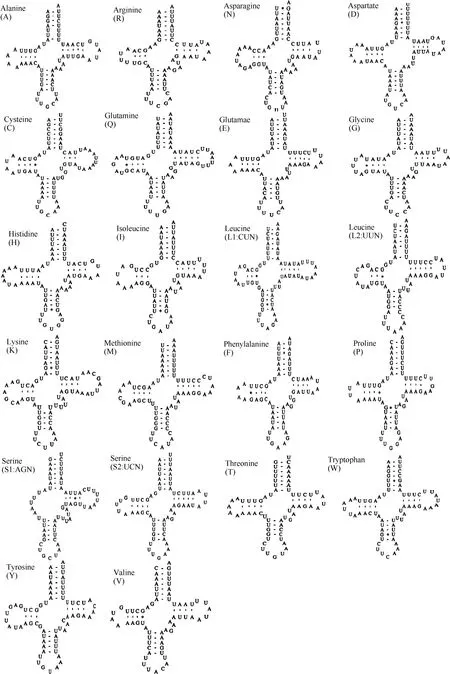
Fig. 2 Predicted secondary cloverleaf structures for the 22 tRNA genes of Apatura ilia
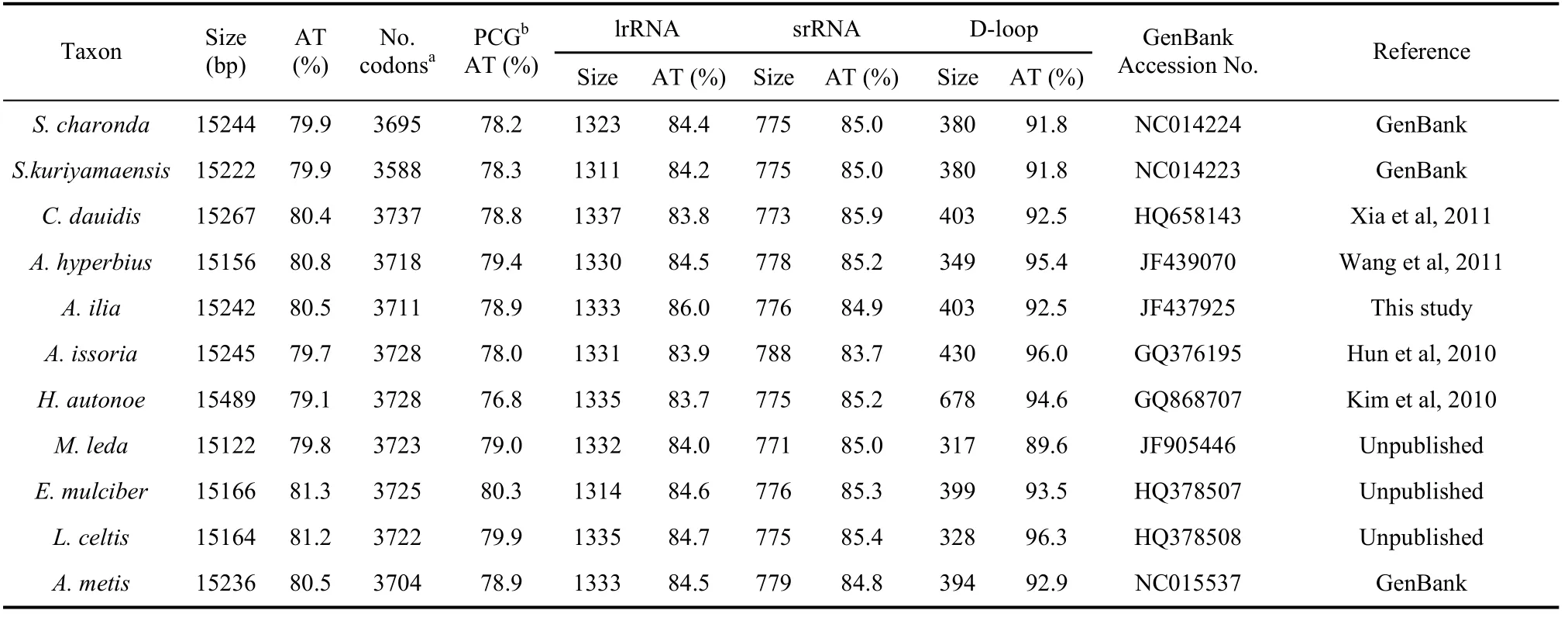
Tab. 2 Comparative characteristics of nymphalid mitogenomes
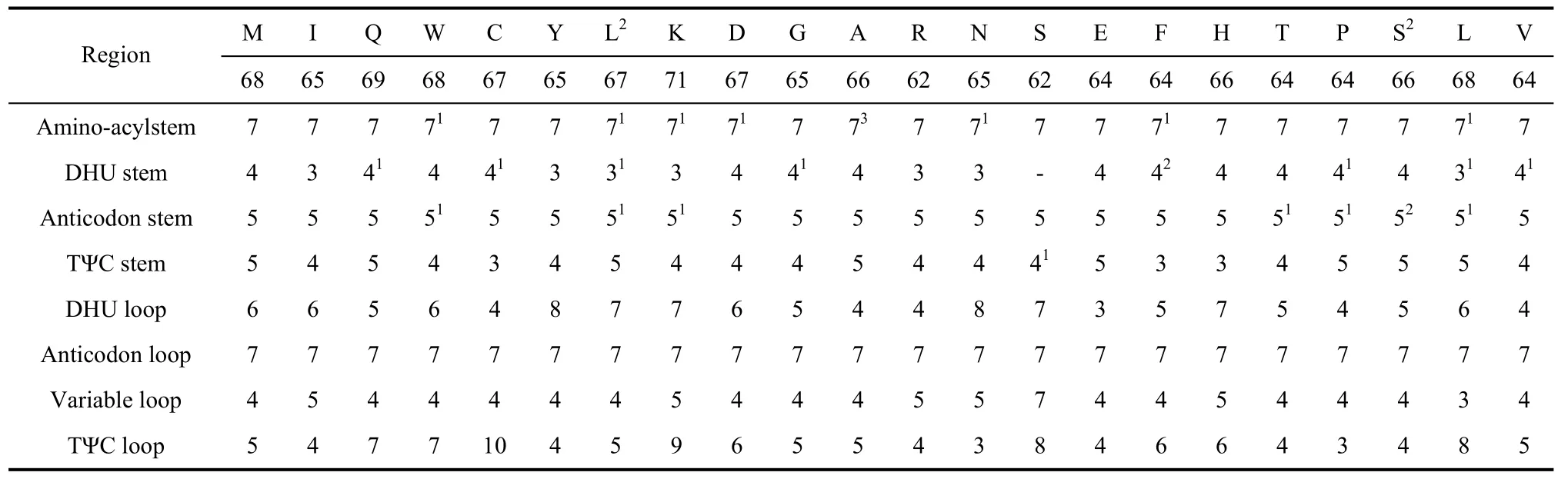
Tab. 3 Sizes of each region of tRNAs in Apatura ilia
3 Discussion
3.1 Gene organization and composition
The length of the complete mitogenome ofA. iliafalls in the known range of the lepidopteran insects from 15 122 bp inM. ledato 16 094 bp inAgehana marho(Papilio maraho) (Wu et al, 2010).A. iliademonstrated a common lepidopteran gene order tRNAMetfollowed by tRNAIleand by tRNAGln, different from those of other insect groups (tRNAIlefollowed by tRNAGlnand by tRNAMet)(Tab. 1). Gene order was used to explore the presumed independent evolutionary lepidopteran lineages after divergence from their common ancestors (Boore et al, 1998).
The nucleotide composition of theA. iliamitogenome showed considerable bias towards an A+T preference (80.5%) (Tab. 2), a common characteristic observed in insect mitochondrial genomes, ranging from 69.5% to 84.9 % (Crozier & Crozier, 1993; Dotson & Beard, 2001). It was noted that the content of base T (40.7%) was slightly higher than base A (39.8%), resulting in an AT skewness value of -0.012. The GC composition (19.5%) was correspondingly lower than AT (Tab. 2) and the GC skewness value was -0.21.
3.2 Protein-coding genes
The putative start codons were found to be the same as in lepidopteran mtDNA (ATN codons: 3 with ATA, 6 with ATG, 3 with ATT) (Tab. 1), except that the COI gene has no uniform start codon (Lessinger et al, 2000; Yukuhiro et al, 2002). In general, lepidopteran insects were quite conservative in using CGA to initiate the COI gene, such as inEriogyna pyretorum(Jiang et al, 2009), Hyphantria cunea(Liao et al, 2010) andAdoxophyes honmai(Lee et al, 2006). However, there are some exceptions. A previous study using transcript information from the cDNA sequence showed that the start codon for the COI gene was TCG (Serine) in dipteran insects (Krzywinski et al, 2006). In addition, TTAAAG has been previously proposed to be the start codon for the COI gene inPieris rapae(Mao et al, 2010), ATTACG forPapilio xuthus(Feng et al, 2010), TTAG forCorean raphaelis(Kim et al, 2006), and TTG forAcraea issoria(Hu et al, 2010) andCalinaga davidis(Xia et al, 2011).
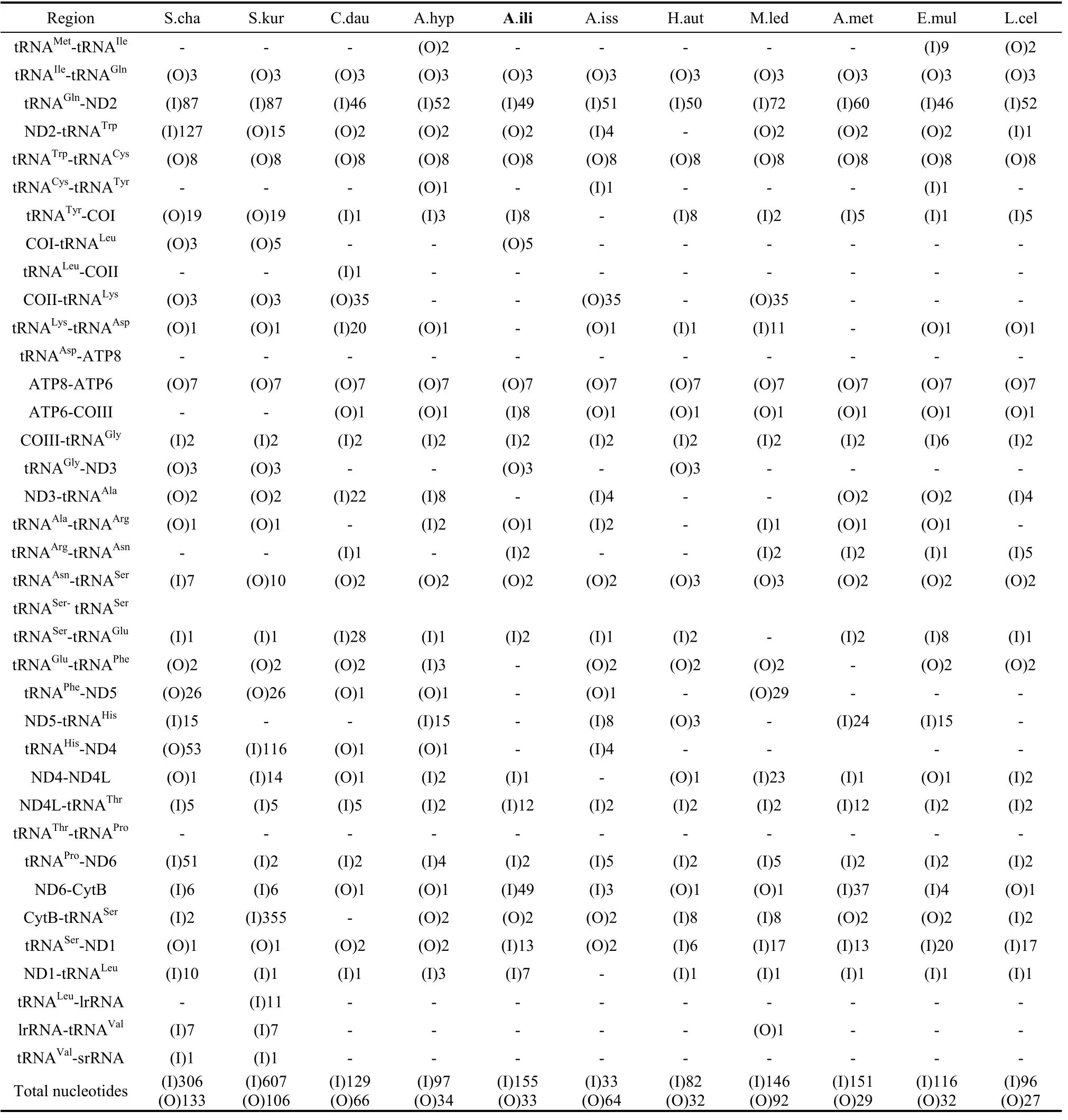
Tab. 4 Overlapping and intergenic spacer sequences of nymphalid mitogenomes
InA. ilia, four genes ended with a single T right ahead of tRNA genes (Tab. 1). The single T residue could be completed into triplet codons by polyadenylation (Clary et al, 1985), and the tRNA secondary structure is functional to the precise cleavage of the mature protein-coding genes from the primary multicistronic transcripts (Ojala et al, 1980, 1981).
The AT bias of the protein-coding genes inA. iliawas prominent with AT content of 78.9%, the same as the average value of sequenced nymphalid mitogenomes (78.9%) (Tab. 2). Additionally, the PCG nucleotide frequency was T>A>G>C, displaying significant skews at AT (–0.15) and GC (0.02), both comparable to other sequenced lepidopterans (Liao et al, 2010). Examination of the concatenated 13 PCGs showed that the third codon position (91.7%) contained higher AT content than the first (74.4%) and second (70.9%) positions, and this case is also similar to other sequenced lepidopteran species likeHapprchia autonoe(Kim et al, 2010). As for A+T content among 13 PCGs, ATP8 had the highest (93.9%) and COI has the lowest (72.4%) values (Tab. 2).
TheA. iliamitogenome also has an AT bias in codon usage. Among 3 711 codons there were 451 UUAs, 439 AUUs, 344 UUUs, 270 AUAs, 229 AAUs and 164 UAUs. These codons have much higher frequencies than others (Tab. 5).
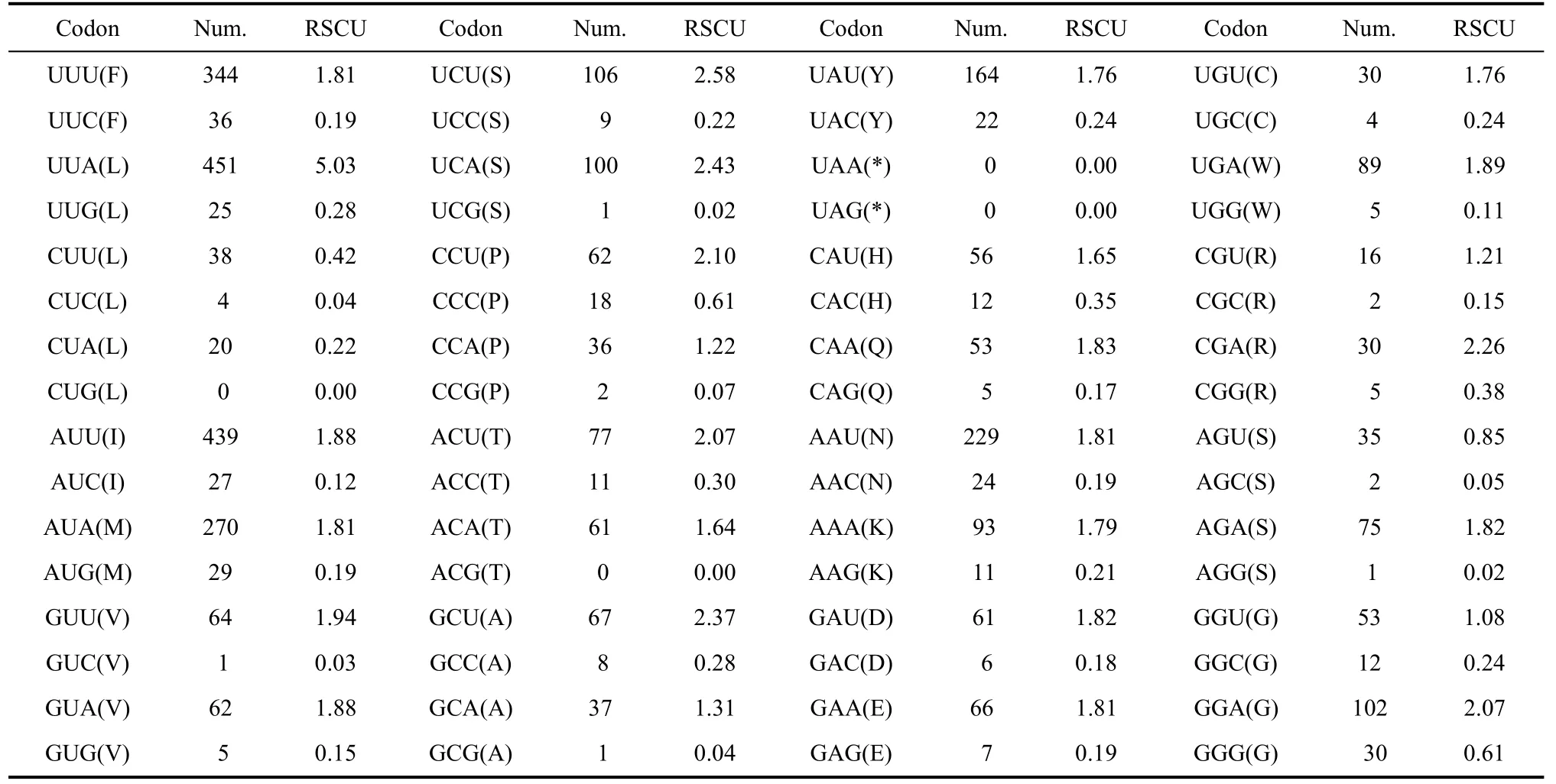
Tab. 5 Codon usage of PCGs in the Apatura ilia mitogenome
3.3 Transfer RNA genes and ribosomal RNA genes
The DHU arm of tRNAser(AGN) has only a simple loop, common in most insects (Hong et al, 2008; Kim et al, 2006; Salvato et al, 2008; Wolstenholme, 1992). Similar toParnassius bremeri(Kim et al, 2009), the tRNAs ofA. iliaharbor 7 base pairs in amino-acyl stems, 5 base pairs in anticodon stems, and 7 base pairs in anticodon loop. However, the base pair numbers vary in other tRNA portions, especially within the TΨC loops (3-10 bp) (Tab. 3).
Among the 22 tRNA genes 32 mismatched base pairs were found, 10 on the amino acyl stem, 9 on the DHU stem, 1 on the TΨC stem and 9 on the anticodon stem. Twenty were between guanine and uracil, justifiable in terms of structural stableness (Topal & Fresco, 1976). Some unconventional mismatches were also observed, e.g. A-C (1), A-G (1), and U-U (8). Similar cases were also seen in other lepidopteran species. For example,C.raphaelishas 8 U-U mismatches in tRNAs;A. issoriaexhibits a G-A and a CU mismatches in the tRNAIle;H. autonoecontains a U-U and a A-C mismatches in tRNALeu(UUR),H. cuneahas U-U mismatches in tRNAAla, tRNALeu(CUN), and tRNALeu(UUR) (Liao et al, 2010). These mismatches can be corrected through RNA-editing mechanisms that are well known for arthropod mtDNA (Lavrov et al, 2000)
As in other lepidopteran species, theA. iliamitogenome was found to harbor two rRNA genes (776 bp srRNA and 1 333 bp lrRNA). The AT content of srRNA was 84.9%, similar to those of other lepidopteran insects (87.5% forPhthonandria atrilineata(Yang et al, 2009), 82.0% forOstrinia furnacalis(Coates et al, 2005)) (Tab.2). The AT content of lrRNA (85.0%) also fell into the range for other lepidopteran insects (85.1% forP. atrilineata, 81.4% forOstrinia Lunifer(Salvato et al, 2008)) (Tab. 2).
3.4 Intergenic spacer and overlapping sequence
The spacer 1 of 49 bp was located between the tRNAGlnand ND2 genes and this spacer had the same size to that inO. furnicalis,Ostrinia nubilalis(Coates et al, 2005),B. moriandB. mandarina(Yukuhiro et al, 2002).It has been suggested that the spacer would remain invariant in length in some congeneric species (Cameron & Whiting, 2008), however, new findings showed divergence amongApatura Metis(unpublished, NC-015537) andA. ilia(Tab. 4). The comparison of this spacer with the neighboring ND2 gene showed 63% homology. Likewise, the cases forH. autonoe(74%),P. bremeri(70%),P. atrilineata(70%),C. raphaelis(62%),A. melete(70%) andB. mori(63%) all suggest that this spacer sequence originated from the ND2 gene (Kim et al, 2009).
Like spacer 1, spacer 2 was also 49 bp in length and found between ND6 and Cyt b. It is notable that a microsatellite-like repeat (TA)23was identified within this region. This case is extremely rare among the known nymphalid mitogenomes (the other example is inA. metiswhich has (TA)12in this region).
Spacer 3 was 13 bp and located between tRNASer(CUN) and ND1. This spacer sequence held a 7-base motif ATACTAA. A similar motif has been identified in previous studies as a plausible conservancy in all Lepidoptera species sequenced so far (Kim et al, 2009; Cameron & Whiting, 2008; Liao et al, 2010; Salvato et al, 2008). It may be functionally essential in the recognition of the mtDNA TERM (the transcription termination peptide). Recent mitogenomic sequence data has demonstrated mixed results in Nymphalidae species. For example, inH. autonoea 6-bp spacer (Kim et al, 2010) was located between tRNASer(CUN) and ND1, and the 7-bp motif ATACTAA located within tRNASer(CUN), whereas inC. dauidis,A. hyperbiusandA. issoris, the two genes were overlapped for 1 or 2 bp and the motif was located at the 3’ end of the ND1 gene.
The 12-bp spacer 4 was located between ND4L and tRNAThr, being longer than those of other nymphalid species. All remaining spacers inA. iliaare less than 10 bp.
We also found two overlapping sequences which are conservative in Lepidoptera, one was 7 bp long and the other 8 bp. The 7-bp sequence was located between ATP8 and ATP6 as ATGATAA, and the 8-bp overlap was located between tRNATrpand tRNACysas AAGCCTTA, These two sequences have also been detected in other lepidopteran insects, such asP. bremeri,H. cuneaandP. atrilineata. Based on data of the presently sequenced lepidopteran insects, the two overlapping sequences were postulated to be conservative across lepidopteran insect taxa. Another 5-bp sequence was located between COI and tRNALeu. The remaining five overlapping sequences range from 1 to 3 in size. So far the 3-bp overlap between tRNAIleand tRNAGlnhas been found in all sequenced Nymphalidae species.
3.5 A+T-rich region
The AT-rich region functional in mtDNA replication and transcription (Taanman, 1999). The origin of the major-strand replication was studied in the AT-rich region in vertebrates (Tapper & Clayton, 1981), followed by the detection on both strands of mtDNA inDrosophilaspecies (Clary & Wolstenholme, 1987; Fauron & Wolstenholmn, 1980). In recent years investigations were expended to the replication origin of the minor-strand in Diptera, Lepidoptera, Coleoptera and Orthoptera, and the results suggest that the replication origin site of mtDNA minor-strand in insects is located before the poly-T structure, which is standing at the 3’end of the AT-rich region (Saito et al, 2005).
The AT-rich region ofA. iliahas some common or similar structural features for lepidopteran insects. It harbors a 21-bp poly-T stretch located 18 bp upstream from srRNA and preceded by a motif ATAGA. The poly-T stretch and the motif composed the origin site for the minor-strand replication, recognizable as a structural signal by regulating proteins (Kim et al, 2009). Additionally, there are two microsatellite-like repeats of (TA)10and (TA)7, which are preceded by a conserved motif ATTTA and located upstream of (TA)10repeat. At the 5’ end of this region there is a 14-bp poly-A structure, shortened to be 10 bp by inserting a single T base. A similar case was found for other nymphalid butterfly species; for example, the poly-A ofA. issorismtDNA was inserted by a guanine. Functionally, the poly-A was assumed to be the replication origin location of the mtDNA major-strand because of its connection with tRNAMet(Kim et al, 2009).
Acknowledgements:We thank ZHU Shun-Yi (College of Life Sciences, Shanxi University, China) for critical comments on an earlier version of this paper.
Avise JC. 1994. Molecular Markers, Natural History and Evolution[M]. New York: Champman & Hall.
Boore JL. 1999. Animal mitochondrial genomes[J].Nucleic Acids Res,27(8): 1767-1780.
Boore JL, Lavrov DV, Brown WM. 1998. Gene translocation links insects and crustaceans[J].Nature,392(6677): 667-668.
Brown WM. 1983. Evolution of animal mitochondrial DNA[M] // Nei M, Koehn RK. Evolution of Genes and Proteins. Sunderland, MA: Sinauer.
Cameron SL, Whiting MF. 2008. The complete mitochondrial genome of the tobacco hornworm,Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths[J].Gene,408(1-2): 112-123.
Chou I. 1998. Classification and Identification of Chinese Butterflies[M]. Zhengzhou: Henan Scientific and Technological Publishing House.
Chou I. 2000. Monographia Rhopalocerorum Sinensium[M]. Zhengzhou: Henan Scientific and Technological Publishing House.
Clary DO, Wolstenholme DR. 1985. The mitochondrial DNA molecule ofDrosophila yakuba: nucleotide sequence, gene organization, and genetic code[J].J Mol Evol,22(3): 252-271.
Clary DO, Wolstenholme DR. 1987.Drosophilamitochondrial DNA: Conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA[J].J Mol Evol,25(2): 116-125.
Clayton DA. 1992. Transcription and replication of animal mitochondrial DNAs[J].Int Rev Cytol, 141: 217-232.
Coates BS, Sumerford DV, Hellmich RL, Lewis LC. 2005. Partial mitochondrial genome sequences ofOstrinia nubilalisandOstrinia furnicalis[J].Int J Biol Sci,1(1): 13-18.
Crozier RH, Crozier YC. 1993. The mitochondrial genome of the honeybeeApis mellifera: complete sequence and genome organization[J].Genetics,133(1): 97-117.
Dotson EM, Beard CB. 2001. Sequence and organization of the mitochondrial genome of the Chagas disease vector,Triatoma dimidiata[J].Insect Mol Biol,10(3): 205-215.
Fauron CMR, Wolstenholmn DR. 1980. Extensive diversity amongDrosophilaspecies with respect to nucleotide sequences within the adenine + thymine-rich region of mitochondrial DNA molecules[J].Nucleic Acids Res,8(11): 2439-2452.
Feng X, Liu DF, Wang NX, Zhu CD, Jiang GF. 2010. The mitochondrial genome of the butterflyPapilio xuthus(Lepidoptera: Papilionidae) and related phylogenetic analyses[J].Mol Biol Rep,37(8): 3877-3888.
Hao JS, Li CX, Sun XY, Yang Q. 2005. Phylogeny and divergence time estimation of Cheilostome bryozoans based on mitochodrial 16S rRNA sequences[J].Chn Sci Bull,50(12): 1205-1211.
Hong MY, Lee EM, Jo YH, Park HC, Kim SR, Hwang JS, Jin BR, Kang PD, Kim KG, Han YS, Kim I. 2008. Complete nucleotide sequence and organization of the mitogenome of the silk mothCaligula boisduvalii(Lepidoptera: Saturniidae) and comparison with other lepidopteran insects[J].Gene,413(1-2): 49-57.
Hu J, Zhang DX, Hao JS, Huang DY, Cameron S, Zhu CD. 2010. The complete mitochondrial genome of the yellow coaster,Acraea issoria(Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event[J].Mol Biol Rep,37(7): 3431-3438.
Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. 2009. Characterization of the complete mitochondrial genome of the giant silkworm moth,Eriogyna pyretorum(Lepidoptera: Saturniidae)[J].Int Biol Sci,5(4): 351-365.
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR. 2006. The mitochondrial genome of the Korean hairstreak,Coreana raphaelis(Lepidoptera: Lycaenidae)[J].Insect Mol Biol,15(2): 217-225.
Kim MI, Baek JY, Kim MJ, Jeong HC, Kim KG, Bae CH, Han YS, Jin BR, Kim I. 2009. Complete nucleotide sequence and organization of the mitogenome of the red-spotted Apollo butterfly,Parnassius bremeri(Lepidoptera: Papilionidae) and comparison with other lepidopteran insects[J].Mol Cells,28(4): 347-363.
Kim MJ, Wan XL, Kim KG, Hwang JS, Kim I. 2010. Complete nucleotide sequence and organization of the mitogenome of endangeredEumenis autonoe(Lepidoptera: Nymphalidae)[J].Afr J Biotechnol,9(5): 735-754.
Krzywinski J, Grushko OG, Besansky NJ. 2006. Analysis of the complete mitochondrial DNA fromAnopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution[J].Mol Phylogenet Evol,39(2): 417-423.
Lavrov DV, Brown WM, Boore JL. 2000. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipedeLithobius forficatus[J].Proc Natl Acad Sci USA,97(25): 13738-13742.
Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. 2006. The mitochondrial genome of the smaller tea tortrixAdoxophyes honmai(Lepidoptera: Tortricidae)[J].Gene,373: 52-57.
Lessinger AC, Martins Junqueira AC, Lemos TA, Kemper EL, Da Silva FR, Vettore AL, Arruda P, Azeredo-Espin AML. 2000. The mitochondrial genome of the primary screwworm flyCochliomyia hominivorax(Diptera: Calliphoridae)[J].Insect Mol Biol,9(5): 521-529.
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang MG, Niu CJ, Liu YQ, Li MG. 2010. The complete mitochondrial genome of the fall webworm,Hyphantria cunea(Lepidoptera: Arctiidae)[J].Int Biol Sci,6(2): 172-186.
Li ZW, Fu YL. 2000. Endangered and protected butterflies in the world[J].J Hainan Norm Univ Nat Sci,5(2): 102-107.
Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence[J].Nucleic Acids Res,25(5): 955-964.
Mao ZH, Hao JS, Zhu GP, Hu J, Si MM, Zhu CD. 2010. Sequencing and analysis of the complete mitochondrial genome ofPieris rapaeLinnaeus (Lepidoptera: Pieridae)[J].Acta Entomol Sin,53(11): 1295-1304.
Mathews DH. 2006. RNA secondary structure analysis using RNA structure[J].Curr Protocol Bioinform,12(6): 1-14.
Ojala D, Merkel C, Gelfand R, Attardi G. 1980. The tRNA genes punctuate the reading of genetic information in human mitochondrial DNA[J].Cell,22(2): 393-403.
Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria[J].Nature,290(5806): 470-474.
Robbins RK. 1982. How many butterfly species[J].News Lepid Soc,1982: 41-42.
Saito S, Tamuea K, Aotsuka T. 2005. Replication origin of mitochondrial DNA in insects[J].Genetics,171(4): 1695-1705.
Salvato P, Simonato M, Battist A, Negrisolo E. 2008. The complete mitochondrial genome of the bag-shelter mothOchrogaster lunifer(Lepidoptera: Notodontidae)[J].BMS Genomics,9(1): 331.
Simon C, Frati F, Bekenbach A, Crespi B, Liu H, Flook P. 1994. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers[J].Ann Entomol Soc Am,87(6): 651-701.
Simons RB, Weller SJ. 2001. Utility and evolution of cytochrome b in insects[J].Mol Phylogenet Evol,20(2): 196-210.
Singh VK, Mangalam AK, Dwivedi S, Naik S. 1998. Primer premier: Program for design of degenerate primers from a protein sequence[J].Biotechnique,24(2): 318-319.
Taanman JW. 1999. The mitochondrial genome: structure, transcription, translation and replication[J].Biochim Biophys Acta,1410(2): 103-123.
Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0[J].Mol Biol Evol,24(8): 1596-1599.
Tapper DA, Clayton DA. 1981. Mechanism of replication of human mitochondrial DNA: localization of the 5’ ends of nascent daughter strands[J].J Biol Chem,256(10): 5109-5115.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Res,25(24): 4876-4882.
Topal MD, Fresco JR. 1976. Complementary base pairing and the origin of substitution mutations[J].Nature,263(5575): 285-289.
Wang XC, Sun XY, Sun QQ, Zhang DX, Hu J, Yang Q, Hao JS. 2011. The complete mitochondrial genome of the laced fritillaryArgyreus hyperbius(Lepidoptera: Nymphalidae)[J].Zool Res,32(5): 465-475.
Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution[J].Int Rev Cytol,141: 173-216.
Wu LW, Lees DC, Yen SH, Lu CC, Hsu YF. 2010. The complete mitochondrial genome of the near-threatened swallowtail,Agehana maraho(Lepidoptera: Papilionidae): evaluating sequence variability and suitable markers for conservation genetic studies[J].Etomol News,121(3): 267-280.
Xia J, Hu J, Zhu GP, Zhu CD, Hao JS. 2011. Sequencing and analysis of the complete mitochondrial genome ofCalinaga davidisOberthür (Lepidoptera: Nymphalidae)[J].Acta Entomol Sin,54(5): 555-565.
Yang L, Wei ZJ, Hong JY, Jang ST, Wen LP. 2009. The complete nucleotide sequence of the mitochondrial genome ofPhthonandria atrilineata(Lepidoptera: Geometridae)[J].Mol Biol Rep,36(6): 1441-1449.
Yukuhiro K, Sezutsu H, Itoh M, Schmizu K, Banno Y. 2002. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth,Bombyx mandarina, and its close relative, the domesticated silkmoth,Bombyx mori[J].Mol Biol Evol,19(8): 1385-1389.
柳紫闪蛱蝶线粒体基因组全序列及与相关蛱蝶类的比较分析
陈 梅1, 田丽丽1, 石庆会1, 曹天文2,*, 郝家胜1,*
(1. 安徽师范大学 生命科学学院分子进化与生物多样性研究室, 安徽 芜湖 241000; 2. 山西省农业科学院 植物保护研究所昆虫研究室, 山西 太原 030031)
该文对柳紫闪蛱蝶Apatura ilia(鳞翅目:蛱蝶科)的线粒体基因组全序列进行了测定, 同时结合其它已知蛱蝶类的相应序列进行了比较分析。结果显示:柳紫闪蛱蝶的线粒体基因组(GenBank accession no.: JF437925)是一个15 242 bp的环状DNA分子, 包含13个蛋白质编码基因、2个rRNA基因 和 22个tRNA基因。13个蛋白编码基因中, 除了COI基因的起始密码子是CGA外, 其余12个蛋白编码基因都具有标准的ATN起始密码子; 柳紫闪蛱蝶与其它已测的 10种蛱蝶在基因定位和排列顺序方面几乎相同, 只是在非编码序列上存在细微的差异, 其核苷酸的构成及密码子使用频率都处于鳞翅目昆虫的范围之内。22个的 tRNA基因中, 除了 tRNASer(AGN)缺少DHU臂, 其余的tRNA基因都显示为典型的三叶草结构。基因组共存在9处基因间重叠区(总长度为33 bp)以及12个基因间隔区(总长为155 bp, 最长间隔是49 bp, 最短的是1 bp)。在ND6和Cyt b间的间隔区中还发现有(TA)23似微卫星结构。与其他蛱蝶类相似, 403 bp的AT富集区包含有ATAGA, ATTTA二个保守模块(一个21 bp的poly-T,一个10 bp的poly-A), 以及二个似微卫星的重复结构((TA)10和(TA)7)。
线粒体基因组; 鳞翅目; 蛱蝶科; 柳紫闪蛱蝶
2011-10-24;接受日期2012-02-20
Q969.42; Q969.439.2; Q754
A
0254-5853-(2012)02-0191-11
date: 2011-10-24; < class="emphasis_bold">Accepted date
date: 2012-02-20
s:This work was supported by grants from the National Science Foundation of China (41172004, 40871034) and partially by the Provincial Key Projects of Natural Science Foundation, Colleges of Anhui Province (KJ2010A142)
* Corresponding author (通信作者),E-mail: ctwen@126.com; jshaonigpas@sina.com
book=192,ebook=139
book=200,ebook=147
