Preliminary analysis of the mitochondrial genome evolutionary pattern in primates
2012-12-25LiangZHAOXingtaoZHANGXingkuiTAOWeiweiWANGMingLI
Liang ZHAO , Xingtao ZHANG Xingkui TAO Weiwei WANG Ming LI ,*
1. Faculty of Biology, Suzhou University, Suzhou Anhui 234000, China;2. Institute of Zoology, the Chinese Academy of Sciences, Beijing 100101, China
Preliminary analysis of the mitochondrial genome evolutionary pattern in primates
Liang ZHAO1,2, Xingtao ZHANG1, Xingkui TAO1, Weiwei WANG1, Ming LI2,*
1.Faculty of Biology,Suzhou University,Suzhou Anhui234000,China;
2.Institute of Zoology,the Chinese Academy of Sciences,Beijing100101,China
Since the birth of molecular evolutionary analysis, primates have been a central focus of study and mitochondrial DNA is well suited to these endeavors because of its unique features. Surprisingly, to date no comprehensive evaluation of the nucleotide substitution patterns has been conducted on the mitochondrial genome of primates. Here, we analyzed the evolutionary patterns and evaluated selection and recombination in the mitochondrial genomes of 44 Primates species downloaded from GenBank. The results revealed that a strong rate heterogeneity occurred among sites and genes in all comparisons. Likewise, an obvious decline in primate nucleotide diversity was noted in the subunit rRNAs and tRNAs as compared to the protein-coding genes. Within 13 protein-coding genes, the pattern of nonsynonymous divergence was similar to that of overall nucleotide divergence, while synonymous changes differed only for individual genes, indicating that the rate heterogeneity may result from the rate of change at nonsynonymous sites.Codon usage analysis revealed that there was intermediate codon usage bias in primate protein-coding genes, and supported the idea that GC mutation pressure might determine codon usage and that positive selection is not the driving force for the codon usage bias.Neutrality tests using site-specific positive selection from a Bayesian framework indicated no sites were under positive selection for any gene, consistent with near neutrality. Recombination tests based on the pairwise homoplasy test statistic supported complete linkage even for much older divergent primate species. Thus, with the exception of rate heterogeneity among mitochondrial genes,evaluating the validity assumed complete linkage and selective neutrality in primates prior to phylogenetic or phylogeographic analysis seems unnecessary.
Mitochondrial genome; Evolutionary pattern; Codon usage bias; Complete linkage; Evolution neutrality; Primates
The mitochondrial genome of vertebrates exhibits several peculiar features including maternal inheritance,the presence of single-copy orthologous genes, a lack of recombination, evolutionary neutrality, and a high mutation rate—characteristics that make it well suited for evolutionary studies (Pesole et al, 1999; Saccone et al,2000). Among all the characteristics and assumptions of the mitochondrial genome, the two most important aspects for explaining the evolutionary process are the presumed freedom from positive Darwinian (adaptive)natural selection and lack of recombination.Mitochondrial proteins are central to the cellular oxidative phosphorylation pathway and are functionally conserved across metazoan phyla (Gray et al, 1999). The importance of these mtDNA protein products supports the hypothesis that some variation in the mtDNA molecule is adaptive. Nonetheless, of the numerous single mtDNA gene studies and several complete mitochondrial genome comparisons published over the past two decades, very few have been able to reject neutrality in favor of positive Darwinian selection(García-Martínez et al, 1998; Rand et al, 1994). Indeed,previous studies on vertebrates indicated that different levels of variability are attributable to1functional constraints and/or slightly deleterious polymorphisms, apattern consistent with near neutrality (Nachman et al,1996; Templeton, 1996).
Recombination breaks down the correlation in genealogical history between different regions of a genome, which may be the incorrect inference of evolutionary history (Schierup & Hein, 2000). The issue of whether recombination occurs in the human mitochondrial genome remains controversial as the necessary enzymes for recombination are in fact present in the mitochondria, and a few paternal mitochondria do penetrate the egg during fertilization (Thyagarajan et al,1996), providing evidence that suggests recombination is possible, at least in humans. Recent broad surveys of animal mitochondrial genomes have also concluded that recombination is widespread (Piganeau et al, 2004;Tsaousis et al, 2005). Another interesting issue about evolutionary patterns of the mitochondrial genome is the codon usage bias in its protein-coding genes. Many studies have analyzed the differences in codon usage between different organisms and between proteins within the same organism (Bulmer, 1991; Kanaya et al, 2001;Sharp et al, 1995) and one of their main conclusions is that there are strong correlations between codon usage and genomic GC content. Moreover, human codon usage may be determined solely by GC content and its isochores composition (Kanaya et al, 2001). Despite suggestive evidence and hypothesis, the relationships between codon usage and the underlying evolutionary constraints are still not fully understood (Prat et al, 2009).
Since the birth of molecular evolutionary analysis,the evolutionary relationships of our own order, primates,have been of central interest. Strangely, no comprehensive and accurate evaluation for the nucleotide substitution pattern(s) of the various mtDNA genes (i.e., tRNA and rRNA genes, synonymous and nonsynonymous positions of protein-coding genes) have been conducted to date.Most primate studies demonstrated mtDNA evolutionary neutrality and no recombination, and generally reported mtDNA still evolves about 5- to 10-fold more rapidly than single-copy nuclear DNA (nDNA) (Brown et al,1982) at an overall nucleotide substitution rate of 1% per million years (Brown et al, 1979; Wilson et al, 1985).
As large libraries of whole-mtDNA genome sequences from primates accumulate, we are able to conduct more sophisticated genome-wide analyses to obtain solid information on the pattern of whole-mtDNA genome evolution in primates while also determining the relative evolutionary rate of each mitochondrial component. Here, we report on the rates and patterns of mtDNA sequence variation and the codon usage in primates and describe the results of tests conducted to detect recombination and selection. These results will not only be of interest in their own right but will also have implications for the use of mtDNA in evolutionary studies of primates.
Materials and Methods
Sequences
54 Mitogenomic sequences from 44 primate species were downloaded from GenBank (Table 1). All 54 sequences were aligned, edited, and compared using the Sequencher 4.5 (Gene Codes Corporation, Ann Arbor,MI). We excluded all gaps and ambiguous alignment sites, resulting in 14,764 bp of sequence from the 13 protein-coding, 2 rRNA, and 21 of the 22 tRNA genes.Protein-coding sequences from the L-strand-encoded genes (ND6 and 8 tRNA genes) were converted into complementary strand sequences. The tRNA-Glu gene and the Control Region were not included because the alignments of those sequences are difficult due to a large number of highly variable sites, insertions, and deletions.
Phylogenetic analyses and sequence diversity
The genealogical relationships between the mitochondrial genomes were analyzed by parsimony using PAUP* (Swofford, 2002). Bootstrapping (Felsenstein,1985) was used to test monophyly. For this study, 1,000 pseudosamples were generated to estimate the bootstrap proportions.
Nucleotide diversity (π) was calculated for all 13 protein-coding, 2 rRNA, and 21 concatenated tRNA genes. Measures of sequence diversity were also calculated by the sliding window method using the DnaSP 4.10 (Rozas et al, 2003). A window (500 bp in length) was moved along the sequences in steps of 50 bp.Patterns of nucleotide diversity for all positions as well as patterns of synonymous and nonsynonymous nucleotide variation were calculated in each window, and the value was assigned to the nucleotide at the midpoint of the window. The gamma parameter alpha (α), which represents the extent of rate heterogeneity among sites,was estimated from the individual data sets with TREEPUZZLE 5.2 (Schmidt et al, 2002) using the 8-categories option.
Codon usage bias
Several parameters related to codon usage bias,such as the codon bias index (Morton, 1993), the effective number of codons (Wright, 1990), and G + C content at second and third positions as well as overall were estimated for each primate mitochondrial proteincoding genes using DnaSP 4.10 (Rozas et al, 2003).Furthermore, to determine whether the compositional changes of the nucleotide content in the primate mitochondrial protein-coding genes are caused by directional mutation pressure or a result of positive selection, we performed a correlation analysis. If the nucleotide bias affects both the synonymous and nonsynonymous sites in protein-coding genes, positive selection could not solely explain this phenomenon,because positive selection should not affect silentnucleotide positions. The correlation analysis of GC content at the second codon position (nonsynonymous mutations related, GC2) and GC content at third position(synonymous mutations related, GCs3) with CG content of all protein codon genes (GCc) were implemented using R 2.6.2 (Ihaka & Gentleman, 1996).
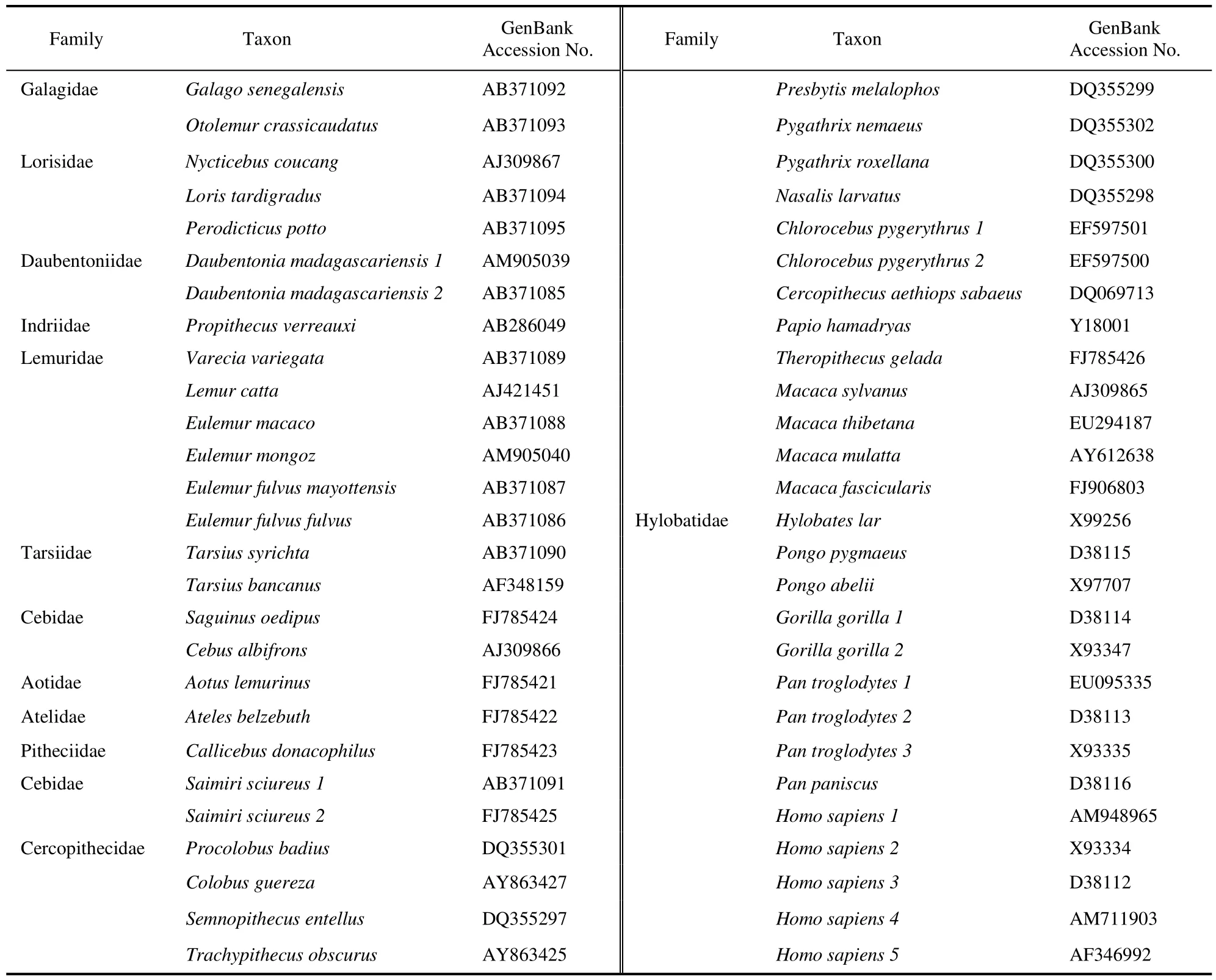
Table 1 Primate taxa and GenBank accession numbers for mitochondria genome genes.
Tests of neutrality
To investigate protein evolution, we first calculated dN/dSratios among the set of 54 primate data and tested their significance usingZ-tests (Nei & Kumar, 2000) for each of the 13 protein-coding genes as implemented by MEGA 4.0 (Tamura et al, 2007). We also tested for sitespecific positive selection with Bayesian posterior probabilities under the Ny98 fitness regime (0<ω1<1,ω2=1, and ω3>1) (Nielsen & Yang, 1998) as described and implemented by MrBayes 3.1.2 (Huelsenbeck &Ronquist, 2001; Ronquist & Huelsenbeck, 2003).
Tests of recombination
We calculated the Kelly (1997) measure of linkage disequilibrium (ZnS); ZnS being the average of R2(Hill& Robertson, 1968) over all pairwise comparisons. We also employed the pairwise homoplasy test (ΦW), a relatively new and robust statistical estimator of recombination (Bruen et al, 2006) that is a coalescentbased estimator of the genealogical correlation or compatibility among sites, which is negatively correlated to recombination. The estimator was calculated, and its significance was estimated via a permutation test using Splitstree 4.10 (Huson & Bryant, 2006).
Results
Phylogenetic analyses and sequence diversity
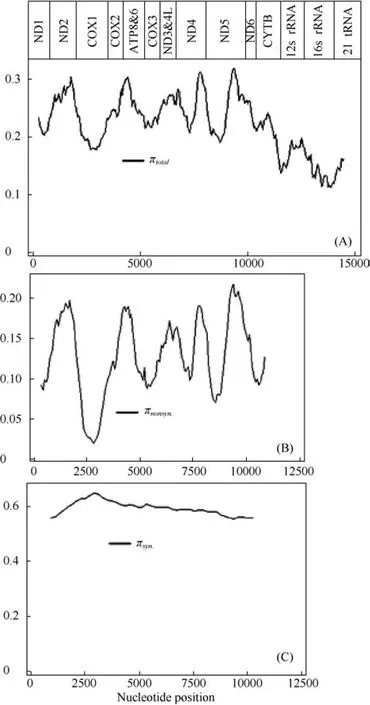
The parsimony tree for primates, based on the mitochondrial genome, is shown in Figure 1. Each internal node was supported by more than 80% bootstrap(data not shown). The mtDNA polymorphism data and estimates of nucleotide divergence are summarized in Table 2, and the distributions of sequence diversityacross the mitochondrial genome are presented in Figure 2. An obvious decline in primate nucleotide diversity was noted in the subunit rRNAs and tRNAs as compared to the protein-coding genes. Among the 13 protein-coding genes, ATP8-ATP6 and the NADH dehydrogenase (ND)complex (with the exception of ND1) showed higher rates of sequence diversity, while the cytochrome oxidase (COX) complex exhibited lower diversity within Primates. Notably, the nonsynonymous divergence plots were similar to those obtained from the total nucleotide divergence, but the trend was not observed in the slidingwindow plot of synonymous divergence. Although the COX complex exhibited an obvious decline in total nucleotide divergence and nonsynonymous divergence,these genes had higher rates of synonymous diversity.Based on the estimated gamma parameter alpha (α), the rate heterogeneity among sites was not even, but the pattern of heterogeneity among sites was distributed across the primate mitochondrial genome.

Figure 1 Parsimony tree of 54 primates based on mitochondrial genome (H1~ H6 were selected nodes for recombination tests)
Codon usage
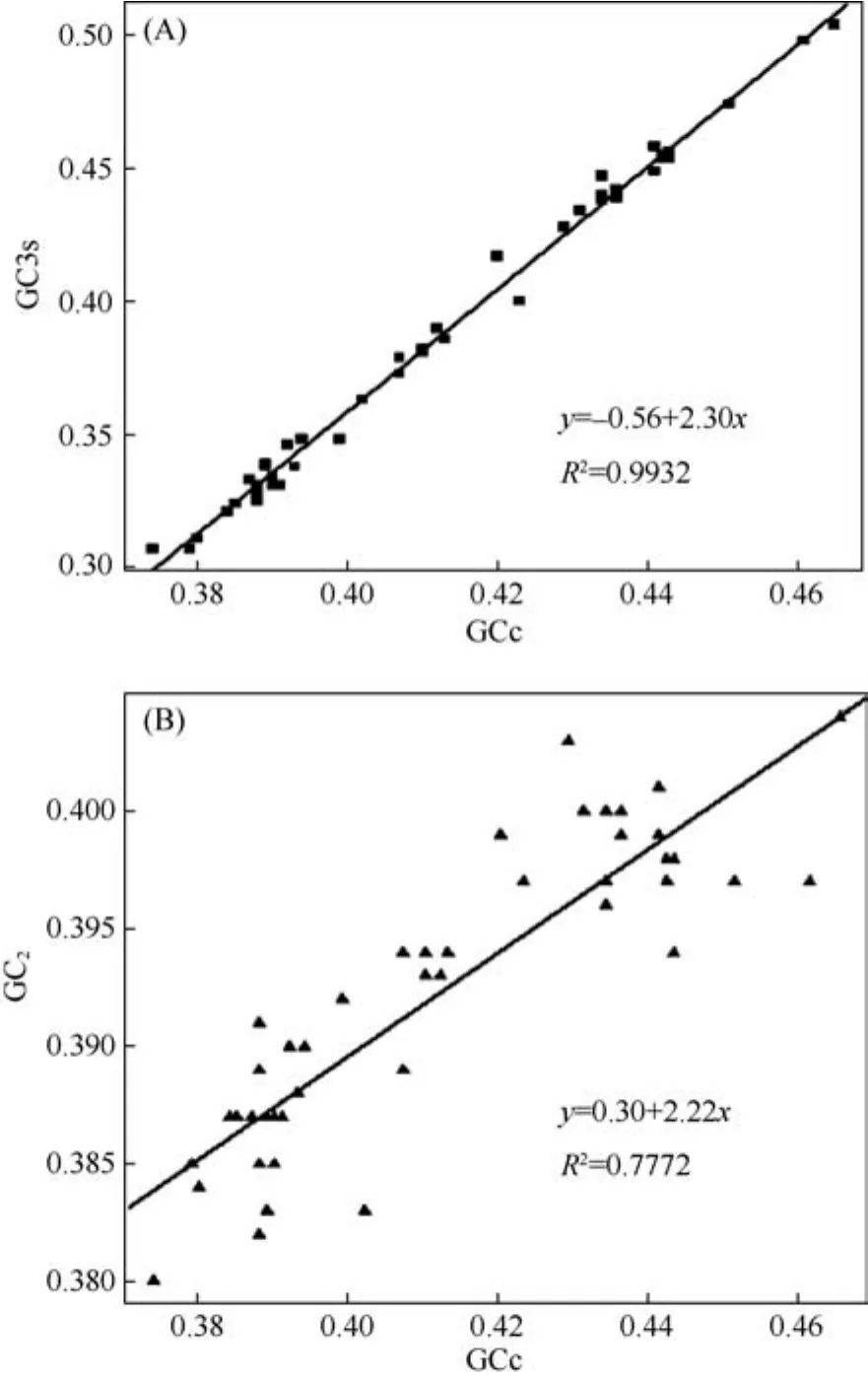
Several parameters related to codon usage bias were estimated to check whether synonymous mutations are selectively neutral. The codon bias index (CBI) is a measure of the deviation from the equal use of synonymous codons with values ranging from 0 (uniform use of synonymous codons) to 1 (maximum codon bias).CBI values were intermediate in our study, ranging from 0.354 to 0.504 (Table 3). The effective number of codons(ENC), which may range from 20 (only one codon is used for each amino acid; i.e., the codon bias is maximum) to 61 (all synonymous codons for each amino acid are equally used; i.e., no codon bias), were likewise intermediate in the primates mitochondrial protein condoning genes (Table 3). GC content at the secondposition (GC2) ranged from 38.0% to 40.4%, GC content at third codon position (GCs3) ranged from 30.7% to 50.4%, and overall CG content of the protein codon genes (GCc) ranged from 37.4% to 46.5%. Correlation analysis indicates that the nucleotide bias affects both the synonymous and nonsynonymous sites in protein-coding genes. Likewise, there is a highly significant correlation between the GC3s and GCc (R2=0.9932, P<0.001;Figure3 A). The correlation between GC2and GCc is also highly significant (R2= 0.7772, P<0.001; Figure 3B).

Table 2 Characteristics for each gene data set in the 54 Mitogenomic sequences from 44 primate species
Neutrality Tests
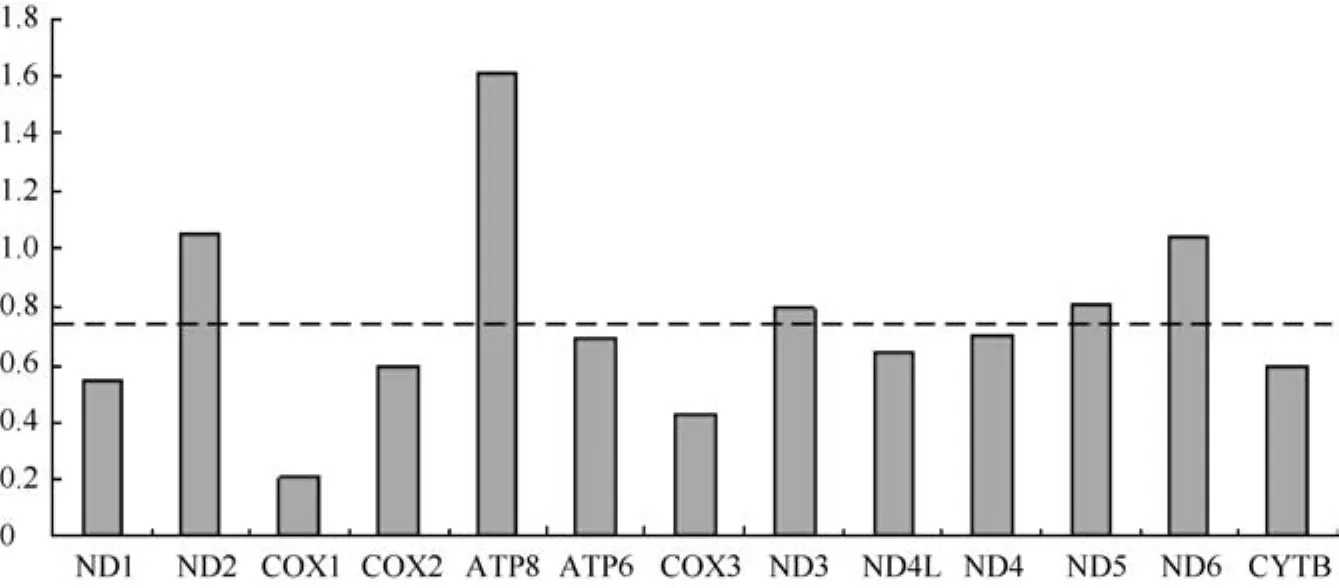
As a conservative measure of selection,Z-tests were conducted for each protein-coding gene in all species. In all cases, dN Table 3 Summary of codon usage index of 13 concatenated protein genes from 44 Primates species Mitogenomic sequences Figure 2 Sequence diversity of the mitochondrial genome by nucleotide position The measures of linkage disequilibrium are summarized in Table 3. The average R2(Hill &Robertson 1968) across all pairwise comparisons (ZnS)for each individual data set and the pooled data provided evidence of recombination between relatively distant divergent species but not for more recent divergent species. The pairwise homoplasy test statistic (ΦW) for the pooled data also provided similar results to the ZnS-test, but the ΦWtest statistic provided no evidence of recombination for each individual data set. Based on our data sets, the assumption of complete linkage of the mtDNA genome appears to be justified only between more recently divergent species. Figure 3 Correlation of GC content at the third codon position(GCs3), and GC content at the second position (GC2)with overall CG content (GCc)(A) Correlation of GCs3 and GCc of coding sequences; (B)Correlation of GC2 and GCc of coding sequences. Primate mitochondrial genomes exhibit striking patterns of both polymorphism and substitution rate heterogeneity among sites and genes, as evidenced by sliding-window estimates of sequence diversity, nonsynonymous and synonymous divergence, and the estimated gamma parameter alpha (α) (Table 2, Figure 2). Less primate nucleotide diversity was observed in the subunit rRNAs and tRNAs than that in the protein oding genes. Among the 13 coding genes, rates were higher in the ND genes and the ATP8- ATP6 gene and lower in the COX complex genes, consistent with the results from various mammals (Kumazawa et al, 2004).Most notably, the nonsynonymous divergence plots were similar to those obtained from the pooled data nucleotide divergence, while the synonymous changes were only slightly different between individual genes of mtDNA(Figure 2). Accordingly, rate heterogeneity may result from the rate of change at nonsynonymous sites. Figure 4 Ratios of nonsynonymous/synonymous rate per gene in the mitochondrial genomes of 54 primates.Dotted line indicates the average ratio. Table 4 Recombination test for primate mitochondrial genomes. The observed pattern of heterogeneity across the mitochondrial genome was similar to the pattern of mitochondrial genome evolution at the intraspecific level in humans (Elson et al, 2004) and both the inter- and intraspecific levels inDrosophila lard(Ballard, 2000),where the synonymous distribution was more homogeneous and the nonsynonymous distribution was more heterogeneous. Additionally, the results obtained here differed from the inter- and intraspecific studies based on whole genome inGadus morhuaand its relative species (Marshall et al, 2009). Interestingly, all mitochondrial genome comparisons mentioned above showed significant rate heterogeneity and site specific hyper-mutability. Possible explanations for rate heterogeneity in the mitochondrial genome include selection pressure, variation in the mutation rate of each gene (involving base composition bias and properties of the L-strand during mtDNA replication) (Marshall et al,2009), and the consequence of neutral mutational effects(Galtier et al, 2006). The detected evolutionary patterns of the mitochondrial genome will influence the choice of mtDNA regions used for phylogeographic studies, since the more variable regions of the genome harbor excess homoplasy relative to the more slowly evolving regions(Galtier et al, 2006). Marshall et al (2009) proposed that the more variable regions of the mitochondrial genome harbor excess homoplasy relative to the more slowly evolving regions, so choosing to examine more slowly evolving regions for evolutionary analysis among species,and even within species, with an increase in the sequence length may be wiser. Based on the primate mitochondrial genome comparison in this study, we suggest that the subunit rRNAs genes, tRNAs genes, and the COX complex genes may be promising sites for future investigations of primate phylogeography. CBIs estimated in this study revealed that there was intermediate codon usage bias in primate protein-coding genes (Table.3). Several evolutionary processes have been postulated as the major factors that determine codon usage. For a number of different organisms, it was suggested that codon usage was best explained by selection for tRNA abundance, gene expression levels,and translational optimization (Duret, 2000). In other cases, the codon usage was explained by mutation rate,mutation preference (Powell & Moriyama, 1997),environmental conditions (Goodarzi et al, 2008),generation time (Subramanian, 2008), and recombination rates (Meunier & Duret, 2004). Although the relationships between codon usage and the underlying evolutionary constraints are still not fully understood,two major paradigms of codon usage have been found in most species (Prat et al, 2009). Natural selection is expected to yield a correlation with codon bias in more highly expressed genes. Alternatively, codon bias could be generated by strictly neutral processes, such as mutational biases, where some mutations occur more than others across the genome by local variations in the base composition (Duret, 2002). Correlation analysis indicates that the nucleotide bias affects both the synonymous and nonsynonymous sites in primate protein-coding genes. There is a highly significant correlation between the GC3s and GCc (R2=0.9932,Figure 3A) and the correlation between GC2and GCc is also highly significant (R2=0.7772, Figure 3B). Because positive selection should not affect silent nucleotide positions, our results support the idea that adaptive evolution is not the driving force for the codon usage bias, indicating that GC mutation pressure may determine codon usage in mitochondrial genomes of different primate species. Using a conservative measure of selection,Z-tests indicated that dN A presumed absence of recombination is another key feature of mtDNA that makes it a useful phylogeographic molecule (Marshall et al, 2009). The uniparental inheritance of mtDNA indicates that recombination should be rare or absent, but a broad survey of 267 published animal mtDNA data sets by Piganeau et al(2004) provided indirect evidence of recombination in more than 14% of cases. Based on the pairwise comparisons (ZnS) and the pairwise homoplasy test statistic (ΦW) for the 13 concatenated protein genes,the assumption of complete linkage of the mtDNA genome appears to be justified only between more recently divergent species. However, no evidence of recombination among the individual genes was detected with the pairwise homoplasy test statistic (ΦW) even among the more divergent species, indicating that recombination is perhaps a result of substitution rate heterogeneity in the primate mtDNA genomes.Additionally, as the lineage divergence increases, the divergence of each individual gene may produce artificial significant ZnS values based on all pairwise comparisons from the pooled data and may lead to false detection of recombination. Thus, it appears unnecessary to consider the effects of recombination on the evolution of mtDNA in primates. Ballard JWO. 2000. Comparative genomics of mitochondrial DNA inDrosophila simulans[J].J Mol Evol,51(1): 64-75. Brown WM, George M Jr, Wilson AC. 1979. Rapid evolution of animal mitochondrial DNA [J].Proc Natl Acad Sci USA,76(4): 1967-1971. Brown WM, Prager EM, Wang A, Wilson AC. 1982. Mitochondrial DNA sequences of primates: tempo and mode of evolution [J].J Mol Evol,18(4): 225-239. Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination [J].Genetics,172(4):2665-2681. Bulmer M. 1991. The selection-mutation-drift theory of synonymous codon usage [J].Genetics,129(3): 897-907. Duret L. 2000. tRNA gene number and codon usage in the C. elegans genome are co-adapted for optimal translation of highly expressed genes [J].Trends Genet,16(7): 287-289. Duret L. 2002. Evolution of synonymous codon usage in metazoans [J].Curr Opin Genet Dev, 12(6): 640-649. Elson JL, Turnbull DM, Howell N. 2004. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection [J].Am J Hum Genet,74(2): 229-238. Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap [J].Evolution,39(4): 783-791. Galtier N, Enard D, Radondy Y, Bazin E, Belkhir K. 2006. Mutation hot spots in mammalian mitochondrial DNA [J].Genome Res,16(2):215-222. García-Martínez J, Castro JA, Ramón MR, Latorre A, Moya A. 1998.Mitochondrial DNA haplotype frequencies in natural and experimental populations ofDrosophila subobscura[J].Genetics,149(3): 1377-1382.Goodarzi H, Torabi N, Najafabadi HS, Archetti M. 2008. Amino acid and codon usage profiles: adaptive changes in the frequency of amino acids and codons [J].Gene,407(1-2): 30-41. Gray MW, Burger G, Lang BF. 1999. Mitochondrial evolution [J].Science,283(5407): 1476-1481. Hasegawa M, Cao Y, Yang Z. 1998. Preponderance of slightly deleterious polymorphism in mitochondrial DNA: nonsynonymous/synonymous rate ratio is much higher within species than between species [J].Mol Biol Evol,15(11): 1499-1505. Hill WG, Robertson A. 1968. Linkage disequilibrium in finite populations [J].Theor Appl Genet, 38(6): 226-231. Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees [J].Bioinformatics,17(8): 754-755. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies [J].Mol Biol Evol,23(2): 254-267. Ihaka R, Gentleman R. 1996. R: A language for data analysis and graphics [J].J Comput Graph Stat,5(3): 299-314. Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. 2001. Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis [J].J Mol Evol,53(4-5): 290-298. Kelly JK. 1997. A test of neutrality based on interlocus associations [J].Genetics,146(3): 1197-1206. Kumazawa Y, Azuma Y, Nishida M. 2004. Tempo of mitochondrial gene evolution: Can mitochondrial DNA be used to date old divergences? [J].Endocytobiosis Cell Res,15(1): 136-142. Marshall HD, Coulson MW, Carr SM. 2009. Near neutrality, rate heterogeneity, and linkage govern mitochondrial genome evolution in Atlantic Cod (Gadus morhua) and other gadine fish [J].Mol Biol Evol,26(3): 579-589. Meunier J, Duret L. 2004. Recombination drives the evolution of GC-content in the human genome [J].Mol Biol Evol,21(6): 984-990. Morton BR. 1993. Chloroplast DNA codon use: evidence for selection at thepsb Alocus based on tRNA availability [J].J Mol Evol,37(3):273-280. Nachman MW, Brown WM, Stoneking M, Aquadro CF. 1996.Nonneutral mitochondrial DNA variation in humans and chimpanzees[J].Genetics,142(3): 953-963. Nei M, Kumar S. 2000. Molecular Evolution and Phylogenetics [M].New York: Oxford University Press. Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene[J].Genetics,148(3): 929-936. Ohta T. 1973. Slightly deleterious mutant substitutions in evolution [J].Nature,246(5428): 96-98. Pesole G, Gissi C, De Chirico A, Saccone C. 1999. Nucleotide substitution rate of mammalian mitochondrial genomes [J].J Mol Evol,48(4): 427-434. Piganeau G, Gardner M, Eyre-Walker A. 2004. A broad survey of recombination in animal mitochondria [J].Mol Biol Evol,21(12):2319-2325. Powell JR, Moriyama EN. 1997. Evolution of codon usage bias inDrosophila[J].Proc Natl Acad Sci USA,94(15): 7784-7790. Prat Y, Fromer M, Linial N, Linial M. 2009. Codon usage is associated with the evolutionary age of genes in metazoan genomes [J].BMC Evol Biol,9(1): 285. Rand DM, Dorfsman M, Kann LM. 1994. Neutral and non-neutral evolution of drosophila mitochondrial DNA [J].Genetics,138(3): 741-756. Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models [J].Bioinformatics,19(12):1572-1574. Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. 2003. DnaSP,DNA polymorphism analyses by the coalescent and other methods [J].Bioinformatics,19(18): 2496-2497. Saccone C, Gissi C, Lanave C, Larizza A, Pesole G, Reyes A. 2000.Evolution of the mitochondrial genetic system: an overview [J].Gene,261(1): 153-159. Schierup MH, Hein J. 2000. Consequences of recombination on traditional phylogenetic analysis [J].Genetics,156(2): 879-891. Schmidt HA, Strimmer K, Vingron M, von Haeseler A. 2002. TREEPUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing [J].Bioinformatics,18(3): 502-504. Sharp PM, Averof M, Lloyd AT, Matassi G, Peden JF. 1995. DNA sequence evolution: the sounds of silence [J].Philos Trans R Soc Lond B Biol Sci,349(1329): 241-247. Subramanian S. 2008. Nearly neutrality and the evolution of codon usage bias in eukaryotic genomes [J].Genetics,178(4): 2429-2432. Swofford DL. 2002. PAUP*. Phylogenetic analysis using parsimony.(*and other methods), Version 4.0b10. Sunderland, Massachusetts:Sinauer Associates. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0 [J].Mol Biol Evol,24(8): 1596-1599. Templeton AR. 1996. Contingency tests of neutrality using intra/interspecfic gene trees: the rejection of neutrality for the evolution of the mitochondrial cytochrome xidase II gene in the hominoid primates [J].Genetics,144(3): 1263-1270. Thyagarajan B, Padua RA, Campbell C. 1996. Mammalian mitochondria possess homologous DNA recombination activity [J].J Biol Chem, 271(44): 27536-27543. Tsaousis AD, Martin DP, Ladoukakis ED, Posada D, Zouros E. 2005.Widespread recombination in published animal mtDNA sequences [J].Mol Biol Evol,22(4): 925-933. Wilson AC, Cann RL, Carr SM, George M, Gyllensten UB, Helm-Bychowski KM, Higuchi RG, Palumbi SR, Prager EM, Sage RD,Stoneking M. 1985. Mitochondrial DNA and two perspectives on evolutionary genetics [J].Biol Linn Soc,26(4): 375-400. Wright F. 1990. The 'effective number of codons' used in a gene [J].Gene,87(1): 23-29. 19 January 2012; Accepted: 27 June 2012 s: This project was supported by the National Basic Research Program of China (973 Program: 2007CB411600), the Natural Science Foundation of China (30630016; 30570292) *Corresponding author, E-mail: lim@ioz.ac.cn
Tests of Recombination
Discussion