蛋白质表面模块划分及其在结合位点预测中的应用
2012-12-11王攀文龚新奇李春华陈慰祖王存新
王攀文 龚新奇 李春华 陈慰祖 王存新
(1北京工业大学生命科学与生物工程学院,北京100124;2清华大学生命科学学院,北京100084)
蛋白质表面模块划分及其在结合位点预测中的应用
王攀文1,§龚新奇2,§李春华1,*陈慰祖1王存新1,*
(1北京工业大学生命科学与生物工程学院,北京100124;2清华大学生命科学学院,北京100084)
蛋白质-蛋白质复合物的结合位点预测是计算分子生物学的一个难题.本文对蛋白质-蛋白质复合物数据集Benchmark 3.0中的双链蛋白质复合物进行了研究,计算了单体的残基溶剂可接近表面积和残基间的接触面积,并据此提出了蛋白质表面模块划分方法.发现模块的溶剂可接近表面积与其内部接触面积的乘积(PSAIA)值能够提供结合位点的信息.在78个双链蛋白质复合物中,有74个体系其受体或配体上具有最大或次大PSAIA值的模块是界面模块.将该方法获得的结合位点信息应用在CAPRI竞赛Target 39的复合物结构预测中取得了较好的结果.本文提出的基于模块的蛋白质结合位点预测方法不同于以残基为基础且仅考虑表面残基的传统预测方法,为蛋白质-蛋白质复合物结合位点预测提供了新思路.
蛋白质结合位点预测;模块划分;溶剂可接近表面积;内部接触面积
1 引言
在后基因组时代,蛋白质结构-功能关系的研究已成为生命科学领域的研究热点.1-4随着结构基因组计划的进行和蛋白质结构解析技术的发展,已有大量蛋白质三维结构被测定.1但由于实验测定有关蛋白质功能方面的信息仍存在诸多困难,发展用理论预测方法研究蛋白质-蛋白质相互作用是目前国际上十分关心的问题.4,5蛋白质结合位点的成功预测将带动分子识别机理、复合物结构预测、蛋白质工程和药物分子设计等相关领域研究的长足进展.所以,蛋白质复合物结合位点预测是蛋白质计算领域最重要的问题之一.3,4,6-9为推动蛋白质结构预测和蛋白质-蛋白质对接技术的发展,欧洲生物信息学研究所(European Bioinformatics Institute, EBI)于2001年开始举办蛋白质复合物结构预测竞赛(Critical Assessment of Prediction of Interaction, CAPRI,网址:http://www.ebi.ac.uk/msd-srv/capri).在竞赛中,竞赛组委会选取尚未发表实验结构数据的蛋白质复合物为竞赛内容,要求参赛者在规定时间内,从蛋白质单体结构出发,用分子对接方法对复合物结构进行预测,然后通过网络提交10个预测结果.10
国际上提出的蛋白质结合位点的预测方法主要包括四大类:基于序列的预测;11,12基于结构的预测;13,14基于理化性质的预测15,16和综合考虑以上信息的预测.17,18基于序列的预测方法认为,蛋白质分子进化重要的残基(如结合位点残基)往往具有较高的序列保守性.因此,这类方法常常通过多序列比对来获得各位点氨基酸残基的保守性,并据此预测蛋白质结合位点.基于结构的预测方法认为,蛋白质某些局域的特定结构是形成一定结合位点的基础,如蛋白质结构的疏水口袋区常常结合底物或抑制剂,由于α螺旋结构的刚性较β折叠更强,所以出现在结合位点的几率较小;由于β转角和无规卷曲结构的高度可变性,它们出现在结合部位的可能性较大.基于理化性质的预测方法表明,蛋白质结合位点区域较其它表面在很多物理化学特性上(如疏水性和极性)都存在明显的统计差异.这类预测常常通过机器学习方法对已知结合位点的蛋白质数据库进行训练,得到一定规律后再进行预测.
目前,大部分结合位点的预测方法是以残基为基础,且仅仅考虑蛋白质表面氨基酸残基的性质,忽略了表面近邻的内部残基的贡献.研究表明,蛋白质分子是一个通过残基间各种相互作用共同维系的复杂系统,蛋白质结合界面的残基往往聚集成簇,19,20其堆积密度相对较高;21界面结构是模块化的,模块内残基的内聚性强,模块间的耦合作用不强;22,23结合界面残基与内部残基间的相互作用包含了界面区域的信息.10鉴于以上研究结果,我们认为在蛋白质-蛋白质结合位点预测中,对表面进行模块划分,且考虑内部残基的作用将能够提高预测成功率.
本文通过对Benchmark 3.0数据库22中的蛋白质-蛋白质复合物进行分析研究,提出了一种能够体现界面残基间内聚性的表面模块划分方法,发现模块的溶剂可接近表面积与其内部接触面积的乘积可以提供蛋白质结合位点的信息.应用这一信息,在CAPRI Target 39复合物结构预测中取得了好的结果.
2 研究体系和方法
2.1 数据集
Benchmark 3.0数据库22中共有124个蛋白质-蛋白质复合物结构,在Benchmark 2.0的基础上增加了40个复合物,包括新增的34个双链复合物.我们以其中的78个双链复合物(表1)为研究对象进行统计分析.这些复合物包含了多种功能类型,如酶/抑制剂、抗原/抗体及其它类型.其单体的残基数范围为29-749.
2.2 蛋白质表面模块划分及参数提取
2.2.1 蛋白质表面模块划分
以蛋白质三维结构中的每个氨基酸残基为中心,将与之有接触的所有残基(包括该中心残基、蛋白质内部残基以及表面残基)划分为一个模块(patch).这里采用基于维里几何的Qcontacts算法来判断两残基是否接触并计算它们之间的接触面积.然后剔除所有的内部模块(不包含任何表面残基的模块),保留表面模块(至少有一个表面残基的模块)作为最后表面模块划分的结果(图1).表面残基定义为相对溶剂可接近面积≥15%的残基,溶剂可接近表面积的计算采用NACCESS(http://www.bioinf.manchester.ac.uk/naccess/)算法,水分子探针半径取0.14 nm.另外,定义界面模块为表面模块中的一部分,且其中至少含有一个与伙伴分子(partner molecule)相互作用的界面残基.界面残基定义为与伙伴分子中至少一个残基有接触的残基.
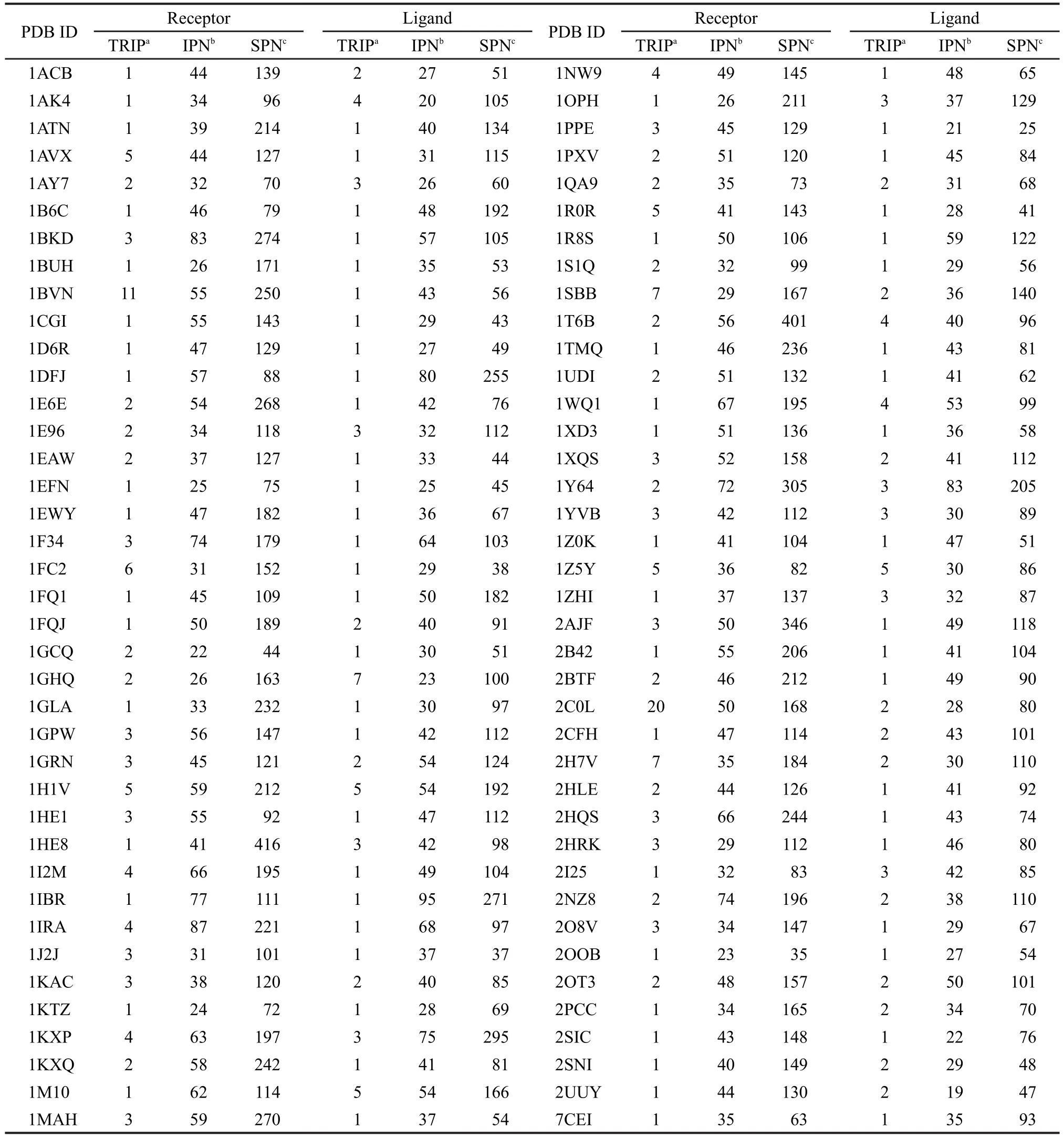
表1 78个蛋白质-蛋白质复合物中单体的模块分析结果Table 1 Patch analysis results of the monomers from 78 protein-protein complexes
2.2.2 表面模块参数的定义
引入两个表面模块参数:溶剂可接近表面积A和内部接触面积C(图1).模块的溶剂可接近表面积A为模块中所有残基的溶剂可接近表面积之和

其中Ai是模块中第i个残基的溶剂可接近表面积.模块的内部接触面积C为模块内所有残基对的接触面积之和

其中Cij是模块中残基i和残基j之间的接触面积.由以上两个参数的乘积可得到模块的PSAIA(溶剂可接近表面积与内部接触面积的乘积)值,即溶剂可接近表面积乘以内部接触面积
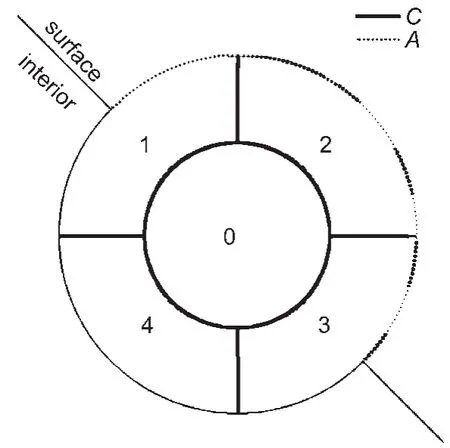
图1 蛋白质表面模块的划分Fig.1 Definition of protein surface patchesResidue 0,which contacts with residues 1,2,3,and 4,is the central residue of this surface patch.Residue 4 is considered as one part of this patch even though it is an internal residue.The dotted lineA stands for the solvent accessible surface area of this patch and the bold solid line C represents the interior contact area.

根据对78个双链复合物中所有单体表面模块PSAIA值的统计分析,确定具有最大或次最大PSAIA值的表面模块为界面模块.
2.3 CAPRI竞赛中Target 39的复合物结构预测
Target 39是CAPRI Round 17中提供的一个复合物结构预测题目,其受体和配体结构由加拿大多伦多大学PARK Hee-Won教授提供(http://www.ebi. ac.uk/msd-srv/capri/round17/round17.html).受体A链有357个氨基酸残基,是蛋白质centaurin-alpha 1,又称3,4,5-三磷酸磷脂酰肌醇(PIP3)结合蛋白,是一种在神经系统中高表达的ADP核糖基化因子(ARF)激活蛋白;B链有98个氨基酸残基,为KIF13B蛋白的叉状(FHA)结构域,KIF13B属于驱动蛋白超家族(KIF).
采用课题组最近发展的HoDock集成分子对接方法24,25对Target 39进行结构预测,基本步骤如下:第一步预测受体和配体的界面模块,用本文提出的预测方法给出蛋白质单体中可能的结合位点区域;第二步采集初始复合物结构,在此过程中,一种方式是用蛋白质可能的结合部位约束复合物模式采集的范围,另一种方式是在全局采样后,把结合位点信息与能量打分综合起来排除错误结构,通过这两种方式挑出少数可能正确的复合物结构;第三步采集精细复合物结构,在上一步结果的基础上做限制搜索范围更加精细的局部对接,此时考虑结合界面上侧链原子的柔性,同时对结构进行打分;第四步根据上面生成的复合物结构之间的相似度,对它们进行成簇聚类;最后综合打分、成簇和结合位点信息挑出10个结构作为最终结果提交给CAPRI竞赛委员会.
3 结果与讨论
3.1 根据表面模块的PSAIA值预测界面模块
对Benchmark 3.0数据集中的78个双链蛋白质复合物中的全部单体进行模块划分,剔除内部模块,仅保留表面模块.计算表面模块的PSAIA值,并将其从大到小排序.表1给出了这些体系的界面模块在全部表面模块中的最高排序、界面模块数及表面模块数.从表1可以看出,对于整个数据集的蛋白质单体,界面模块数的范围是19-95,表面模块数的范围为25-416,有150个单体的界面模块在全部表面模块中的最高排序在5以内(包括5),占全部单体的96.15%;有60个复合物的受体或配体上具有最大PSAIA值的表面模块是界面模块,占整个数据集的76.92%;有74个复合物的受体或配体上具有最大或次大PSAIA值的表面模块是界面模块,占整个数据集的94.87%;20个复合物的受体和配体的具有最大PSAIA值的模块都是界面模块.以上结果说明蛋白质单体中具有最大或次大PSAIA值的表面模块倾向于参与蛋白质-蛋白质相互作用.换言之,即内部接触紧密且对外暴露充分的表面模块(表现为模块内部接触面积C与其溶剂可接近表面积A的乘积大)易出现在界面上,这在一定程度上体现了前人的观点:蛋白质结合界面残基往往聚集成簇,19,20堆积密度相对较高;21界面结构是模块化的,模块内残基的内聚性强;22,23结合界面残基与内部残基间的相互作用包含了界面区域的信息.10进一步仔细分析发现,这种倾向性对于受体和配体是有所不同的.在全部单体中,具有最大PSAIA值的表面模块是界面模块的受体数为33个,而具有这种情况的配体数是47个;具有最大或次大PSAIA值的模块是界面模块的受体数为50个,而具有这种情况的配体数是62个.这在一定程度上说明配体更倾向于拥有这种特性.这一点可作如下解释:配体分子大多是较小的球形蛋白,受体分子大多是较大的不规则蛋白,结合界面常常呈凸形突出于表面,这样与界面残基接触的内部残基数相对于与凹陷的非界面残基接触的内部残基数较少,导致界面模块的内部接触面积C变小,其PSAIA值也随之变小,结果界面模块就不容易被排到最前面.根据以上分析,得出拥有最大或次大PSAIA值的表面模块易出现在界面上,因此可以利用这一性质来预测蛋白质的界面区域.
3.2 具体实例分析
图2(a)和2(b)显示了两个蛋白质复合物1ATN和2C0L的单体上具有最大或次大PSAIA值的模块在分子表面的位置.复合物1ATN由A链受体和D链配体组成,氨基酸残基数分别是372和258;复合物2C0L由A链受体和B链配体组成,氨基酸残基数分别是292和122.从图2(a)看出,对于1ATN的两个单体,其具有最大PSAIA值的模块都是界面模块.在该复合物中,受体和配体的形状较为规则,特别是配体,更接近球形,界面模块的预测相对容易.图2(b)显示,复合物2C0L的界面是由配体的一个柔性较大的无规卷曲结构插入受体的由多个α螺旋组成的腔洞形成的.除无规卷曲结构外,配体的整体形状接近球形,具有次大PSAIA值的模块在界面上.对于受体而言,它的结合部位包含了由多个α螺旋组成的腔洞,造成其内部残基的堆积比较松散,界面模块的PSAIA值不高,使得界面模块预测错误.
3.3 CAPRI竞赛中Target 39的结构预测
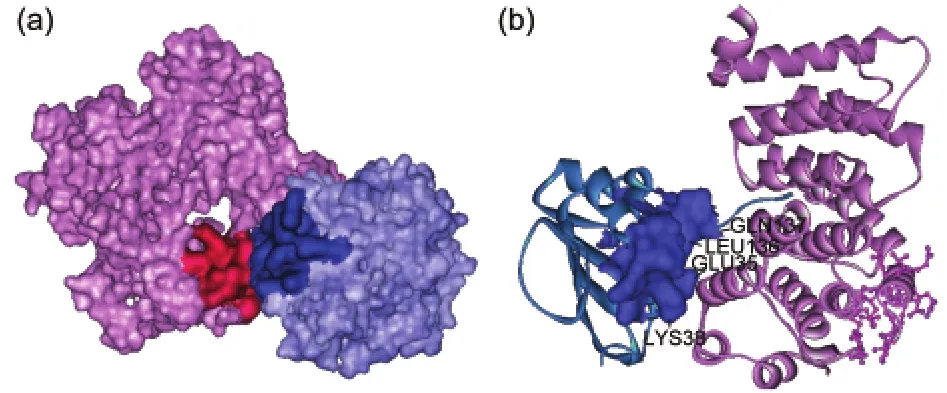
图2 蛋白质-蛋白质复合物中单体上具有最大或次大PSAIA值的模块所在的表面区域Fig.2 Surface areas of the patches with the first or second greatest product of the solvent accessible area and the interior contact area(PSAIA)value on monomers of protein-protein complexes(a)complex 1ATN.The pink monomer is the receptorAchain and the blue one is the ligand D chain.The deep pink and deep blue labeled surface areas are the interface patches with the first greatest PSAIAvalues.(b)complex 2C0L.The pink monomer is the receptor Achain and the blue one is the ligand B chain.The deep blue area of ligand is the interface patch with the second greatest PSAIAvalue, and the interface residues are labeled out.The receptor pink residues displayed by ball and stick compose the patch with the first greatest PSAIA,which is not an interface patch.
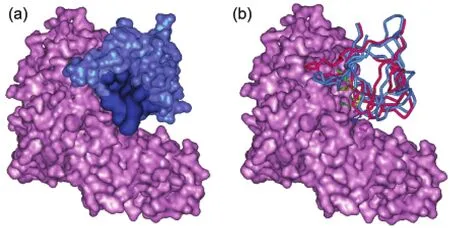
图3 Target 39配体界面模块预测和复合物结构预测结果Fig.3 Results on interface patch prediction of ligand and the complex structure prediction for Target 39(a)interface patch prediction of ligand.The pink monomer is the receptorAchain and the blue one is the ligand B chain.The deep blue area is the predicted interface patch with the first greatest PSAIAvalue,which really interacts with the receptor. (b)Superposition of the predicted best structure and the corresponding X-ray complex structure.The ligand in X-ray structure is drawn in red tubes,and the docked ligand in blue tubes.The interface residues ASN452,MET486,LEU533,ASN536,ASN537,and PHE539 are marked by ball and stick and colored in green,pink,yellow, blue,deep green,and brown,respectively.
从以上分析可以看出,用蛋白质表面模块的PSAIA值来预测形状较规则的球形蛋白的结合部位效果比较好.图3显示了CAPRI Target 39复合物结构预测与配体界面模块预测结果.由图可见,配体接近球形,受体不规则.因此,为了避免结构预测错误,在分子对接中,仅考虑配体的界面模块预测信息,而未考虑受体的预测结果(实际上受体具有最大PSAIA值的模块不在结合界面上).在图3(a)中,配体最大PSAIA值模块的表面被标记为深蓝色.该模块中的表面残基有9个:ASN452、CYS484、GLY485、MET486、LEU533、ASN536、ASN537、HIS538和PHE539,其中真正参与相互作用的界面残基有6个:ASN452、MET486、LEU533、ASN536、ASN537和PHE539.这说明我们提出的识别蛋白质界面模块的方法可比较准确地预测配体的结合位点.最后综合打分、成簇和结合位点信息挑出10个结构作为最终结果提交给CAPRI组委会.国际上有37个小组参加了该复合物的结构预测,共提交了366个结构,其中只有3个结构达到了CAPRI组委会制定的“好结构”的标准.26我们提交的一个结构为其中之一,其配体主链原子均方根偏差(L_rmsd)为0.25 nm(图3(b)),被评为中等(Medium)好结构(http://www.ebi.ac.uk/msd-srv/capri/round17/round17. html).HoDock分子对接方法以及配体结合位点正确信息的获取为该结构的成功预测提供了重要保障.
4 结论
通过对蛋白质-蛋白质复合物数据集Bench-mark 3.0中的双链复合物分析研究,提出了蛋白质表面模块的划分方法,并发现模块的溶剂可接近表面积与其内部接触面积的乘积值PSAIA,能够提供结合位点信息,从而建立了界面模块预测方法.用该方法预测形状较规则的球形蛋白的结合部位效果较好.将该方法获得的结合位点信息应用在CAPRI竞赛Target 39的复合物结构预测中取得了较好的结果.传统的结合位点预测方法大都是以残基为基础,且仅仅考虑表面残基.本文提出的基于模块的蛋白质结合位点预测方法不同于传统方法,不仅考虑了表面残基,而且考虑了内部残基对的贡献,并以模块为基础来预测结合位点,这为蛋白质-蛋白质相互作用中结合位点预测方法研究提供了新的思路.
(1) Teichmann,S.A.;Murzin,A.G.;Chothia,C.Curr.Opin.Struct. Biol.2001,11(3),354.doi:10.1016/S0959-440X(00)00215-3
(2) Baker,D.;Sali,A.Science 2001,294(5540),93.doi:10.1126/ science.1065659
(3) Stark,A.;Shkumatov,A.;Russell,R.B.Structure 2004,12(8), 1405.doi:10.1016/j.str.2004.05.012
(4) Jones,S.;Thornton,J.M.Curr.Opin.Chem.Biol.2004,8(1), 3.doi:10.1016/j.cbpa.2003.11.001
(5) Kinoshita,K.;Nakamura,H.Curr.Opin.Struct.Biol.2003,13 (3),396.doi:10.1016/S0959-440X(03)00074-5
(6)Tseng,Y.Y.;Li,W.H.Proc.Natl.Acad.Sci.U.S.A.2011,108 (13),5313.doi:10.1073/pnas.1102210108
(7)Amos-Binks,A.;Patulea,C.;Pitre,S.;Schoenrock,A.;Gui,Y.; Green,J.R.;Golshani,A.;Dehne,F.BMC Bioinformatics 2011, 12,225.doi:10.1186/1471-2105-12-225
(8) Xiong,Y.;Liu,J.;Wei,D.Q.Proteins 2011,79(2),509.doi: 10.1002/prot.v79.2
(9) He,X.;Chen,C.C.;Hong,F.;Fang,F.;Sinha,S.;Ng,H.H.; Zhong,S.PLoS One 2009,4(12),e8155.
(10) Lensink,M.F.;Mendez,R.;Wodak,S.J.Proteins 2007,69(4), 704.doi:10.1002/prot.21804
(11) Lichtarge,O.;Bourne,H.R.;Cohen,F.E.J.Mol.Biol.1996, 257(2),342.doi:10.1006/jmbi.1996.0167
(12) Ofran,Y.;Rost,B.FEBS Lett.2003,544(1-3),236.doi: 10.1016/S0014-5793(03)00456-3
(13)Laskowski,R.A.;Luscombe,N.M.;Swindells,M.B.; Thornton,J.M.Protein Sci.1996,5(12),2438.
(14) Torrance,J.W.;Bartlett,G.J.;Porter,C.T.;Thornton,J.M.J. Mol.Biol.2005,347(3),565.doi:10.1016/j.jmb.2005.01.044
(15) Gao,Y.;Wang,R.;Lai,L.J.Mol.Model.2004,10(1),44.doi: 10.1007/s00894-003-0168-3
(16) Innis,C.A.;Anand,A.P.;Sowdhamini,R.J.Mol.Biol.2004, 337(4),1053.doi:10.1016/j.jmb.2004.01.053
(17) de Vries,S.J.;Bonvin,A.M.Bioinformatics 2006,22(17), 2094.doi:10.1093/bioinformatics/btl275
(18) Madabushi,S.;Yao,H.;Marsh,M.;Kristensen,D.M.;Philippi, A.;Sowa,M.E.;Lichtarge,O.J.Mol.Biol.2002,316(1),139. doi:10.1006/jmbi.2001.5327
(19) Guharoy,M.;Chakrabarti,P.Proc.Natl Acad.Sci.U.S.A. 2005,102(43),15447.doi:10.1073/pnas.0505425102
(20) Li,X.;Keskin,O.;Ma,B.;Nussinov,R.;Liang,J.J.Mol.Biol. 2004,344(3),781.doi:10.1016/j.jmb.2004.09.051
(21) Hintze,A.;Adami,C.Plos Comput.Biol.2008,4(2),e23.
(22) Hwang,H.;Pierce,B.;Mintseris,J.;Janin,J.;Weng,Z.Proteins 2008,73(3),705.doi:10.1002/prot.22106
(23) Bai,H.J.;Lai,L.H.Acta Phys.-Chim.Sin.2010,26,1988. [白红军,来鲁华.物理化学学报,2010,26,1988.]doi:10.3866/ PKU.WHXB20100725
(24)Gong,X.Q.;Liu,B.;Chang,S.;Li,C.H.;Chen,W.Z.;Wang, C.X.Sci.China Life Sci.2010,53(9),1152.doi:10.1007/ s11427-010-4050-0
(25)Gong,X.Q.;Wang,P.W.;Yang,F.;Chang,S.;Liu,B.;He,H. Q.;Cao,L.B.;Xu,X.J.;Li,C.H.;Chen,W.Z.;Wang,C.X. Proteins 2010,78(15),3150.doi:10.1002/prot.v78:15
(26) Janin,J.;Henrick,K.;Moult,J.;Eyck,L.T.;Sternberg,M.J.; Vajda,S.;Vakser,I.;Wodak,S.J.Proteins 2003,52(1),2. doi:10.1002/(ISSN)1097-0134
May 25,2012;Revised:August 16,2012;Published on Web:August 16,2012.
Division of Protein Surface Patches and Its Application in Protein Binding Site Prediction
WANG Pan-Wen1,§GONG Xin-Qi2,§LI Chun-Hua1,*CHEN Wei-Zu1WANG Cun-Xin1,*
(1College of Life Science and Bioengineering,Beijing University of Technology,Beijing 100124,P.R.China;2School of Life Sciences,Tsinghua University,Beijing 100084,P.R.China)
Binding site prediction for protein-protein complexes is a challenging problem in the area of computational molecular biology.Using a set of double-chain complexes in Benchmark 3.0,we calculated the solvent accessible surface areas and inter-residue contact areas for each monomer and propose a division method of protein surface patches.We found that the products of the solvent accessible surface areas and internal contact areas of patches,the PSAIA values,could provide protein binding site information.In a dataset of 78 complexes,either receptors or ligands of 74 complexes had interface patches with the first or second greatest PSAIA values among all surface patches.A good docking result was achieved when the binding site information obtained with this method was applied in Target 39 of the CAPRI experiment.This patch-based protein binding site prediction method differs from traditional methods,which are based on single residue and consider only surface residues.This provides a new method for binding site prediction in protein-protein interactions.
Protein binding site prediction;Patch division;Solvent accessible surface area; Interior contact area
10.3866/PKU.WHXB201208162
∗Corresponding authors.LI Chun-Hua,Email:chunhuali@bjut.edu.cn.WANG Cun-Xin,Email:cxwang@bjut.edu.cn;Tel:+86-10-67392724.
§These authors contribute equally to this work.The current address of WANG Pan-Wen is Department of Biochemistry,Li Ka Shing Faculty of Medicine,the University of Hong Kong,Hong Kong,P.R.China.
The project was supported by the National Natural Science Foundation of China(31171267,10974008),Beijing Natural Science Foundation,China (4102006),International Science&Technology Cooperation Program of China(2010DFA31710),and Fundamental Research Fund for the Beijing Municipal Education Commission Science and Technology Innovation Platform,China.
国家自然科学基金(31171267,10974008),北京市自然科学基金(4102006),科技部国际合作项目(2010DFA31710)和北京市教委科技创新平台-自然基础研究项目资助
O641