HPLC法测定香桂化浊胶囊中大黄素的含量Δ
2012-12-03刘红淼杨继章李艳玲河北医科大学第一医院石家庄050031石药集团中奇制药技术石家庄有限公司石家庄050051
刘红淼,杨继章,李艳玲(1.河北医科大学第一医院,石家庄050031;.石药集团中奇制药技术(石家庄)有限公司,石家庄 050051)
香桂化浊方为河北医科大学第一医院临床应用多年的经验方,由广藿香、土大黄、肉桂等药材组成。其疗效确切、可靠,具有芳香醒脾、清化湿浊之功,主治因各种疾病后邪留未尽、脾胃内伤所致长期大便溏泄或有黏液、腹胀肠鸣、纳呆,低热,舌苔白腻或白滑或见微黄,脉濡缓;主要用于慢性肠炎、真菌感染致肠炎证属脾虚湿困的治疗。关于本方制剂工艺及质量控制方法的研究已有报道[1~4]。为了更加全面、有效地控制其质量,笔者采用高效液相色谱(HPLC)法对香桂化浊胶囊中土大黄中大黄素的含量进行了测定。
1 仪器与试药
LC-10 A系列HPLC仪,包括LC-10 Avp高压泵、SPD-10 Avp紫外检测器、SCL-10 Avp系统控制器及Class-vp 6.10色谱工作站(日本岛津公司);电子天平(德国Sartorius公司)。
香桂化浊胶囊(批号:20100601、20100602、20100603)和阴性样品均为河北医科大学第一医院制剂室制备;大黄素对照品(中国食品药品检定研究院,批号:110756-200110,供含量测定用);甲醇、乙腈为色谱纯,其他试剂均为分析纯。
2 方法与结果[5,6]
2.1 色谱条件
色谱柱:Hypersil C18(150 mm×4.6 mm,5 μm);流动相:0.1 mol·L-1磷酸二氢钠溶液(用磷酸调pH值至3.0)-甲醇-乙腈(30∶35∶35);流速:1.2 mL·min-1;进样量:20 μL;检测波长:289 nm。
2.2 对照品溶液的制备
精密称取大黄素对照品10.04 mg,置200 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得。
2.3 供试品溶液的制备
取胶囊内容物,精密称取约0.6 g,置100 mL圆底烧瓶中,加0.5mol·L-1盐酸30mL,(80±5)℃水浴2h,放至室温,过滤,滤液用氯仿(20、20、20、10、10 mL)萃取5次,合并萃取液,备用。滤渣晾干,置索氏提取器中,用氯仿提至无色(提取约4.5 h)。与上述萃取液合并,蒸除氯仿,挥干,残渣加甲醇溶解并转移至25 mL量瓶中,定容,摇匀,用0.45 μm微孔滤膜滤过,即得。
2.4 专属性试验
取不含土大黄的阴性样品,按“2.3”项下方法制备阴性对照溶液。取对照品溶液、供试品溶液、阴性对照溶液各适量,按上述色谱条件进样测定。结果表明,阴性对照在大黄素保留时间处无干扰峰出现。色谱见图1。
2.5 线性关系考察
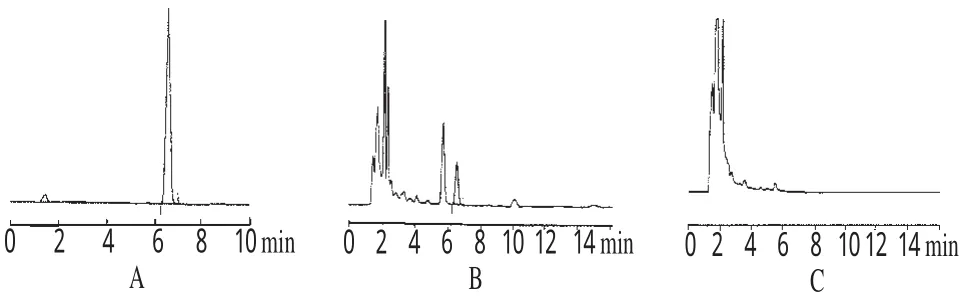
图1 高效液相色谱图Fig 1 HPLC chromatograms
精密量取大黄素对照品溶液0.5、1.0、2.0、3.0、4.0、5.0 mL,分别置于10 mL量瓶中,用甲醇稀释至刻度,摇匀,得系列标准溶液,分别精密量取20 μL,注入HPLC仪,记录色谱图,测定峰面积。以峰面积积分值(Y)对对照品浓度(X)进行线性回归,得回归方程为Y=6.813×104X-1.456×103(r=0.9999,n=6)。结果表明,大黄素的检测浓度在2.51~25.10 μg·mL-1范围内与峰面积积分值呈良好线性关系。
2.6 精密度试验
精密量取同一份对照品溶液(15.6 μg·mL-1)20 μL,连续进样5次,记录峰面积。结果,RSD=0.65%(n=5),表明仪器精密度良好。
2.7 稳定性试验
取同一份供试品溶液,分别于0、2、4、8、12 h进样测定,每次20 μL。结果,RSD=0.73%(n=5),表明供试品溶液在12 h内稳定性良好。
2.8 重复性试验
取同一批(批号:20100602)样品适量,按“2.3”项下方法平行制备6份供试品溶液,用甲醇转移至25mL量瓶中,经0.45 μm微孔滤膜滤过,取续滤液20 μL,注入HPLC仪,测定大黄素含量。结果,样品平均含量为 0.3973mg·g-1,RSD=1.41%(n=6),表明该方法重复性良好。
2.9 加样回收率试验
取已知大黄素含量的香桂化浊胶囊样品(批号:20100602)约0.39 g,精密称定9份,分别精密加入不同量的大黄素对照品溶液,照“2.3”项下方法制备供试品溶液并测定含量,计算加样回收率,结果见表1。
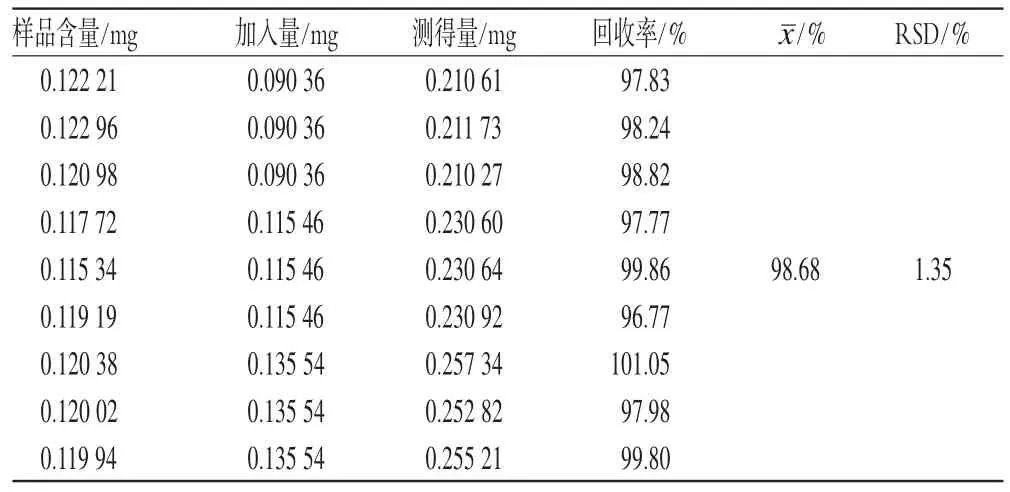
表1 加样回收率试验结果(n=9)Tab 1 Results of recovery test(n=9)
2.10 样品含量测定
取3批样品(批号:20100601、20100602、20100603)各适量,分别按“2.3”项下方法制备供试品溶液,照上述方法测定大黄素的含量。结果,3批样品中大黄素含量分别为0.3941、0.4351、0.3997 mg·g-1。根据此结果,同时考虑药材来源及制剂生产、贮藏等因素,暂定大黄素含量限度为不得少于0.30 mg·g-1。
3 讨论
根据大黄素的理化性质,笔者分别采用3种方法提取同一批样品中的大黄素:(1)超声提取法:样品加50 mL甲醇超声30 min,放冷,过滤,滤液加30 mL 0.5 mol·L-1的盐酸,(80±5)℃水浴水解2 h,放至室温,用氯仿(20、20、20、10、10 mL)萃取至无色,合并萃取液,挥干氯仿,残渣加甲醇溶解并转移至25 mL量瓶中,定容,摇匀,用0.45 μm微孔滤膜滤过,即得。(2)水解、提取同时进行法:样品加0.5 mol·L-1的盐酸30 mL、氯仿30 mL,置(80±5)℃水浴上回流2 h,放至室温,分取氯仿层,水层分别用氯仿(20、20、20、10、10 mL)萃取至无色,合并氯仿层,蒸除氯仿,残渣加甲醇溶解并转移至25 mL量瓶中,定容,摇匀,用0.45 μm微孔滤膜滤过,即得。(3)索氏提取法:将胶囊内容物置100 mL圆底烧瓶中,加入0.5 mol·L-1的盐酸30 mL,置(80±5)℃水浴上水解2h,放至室温,过滤,滤液用氯仿(20、20、20、10、10 mL)萃取5次,合并萃取液,备用;滤渣阴干,置索氏提取器中用氯仿提至无色(约4.5 h),再与上述萃取液合并,蒸除氯仿,挥干,残渣加甲醇溶解并转移至25 mL量瓶中,定容,摇匀,用0.45 μm滤膜滤过,即得。通过测定3种方法所提样品中的大黄素含量,结果表明,方法(2)的色谱图上,杂质峰与大黄素峰难以分离,不能定量;方法(1)的色谱图上供试品杂质峰多,且在过滤过程中易造成不平行操作;而采用方法(3),大黄素提取较完全且杂质峰少、分离效果好、稳定,故采用方法(3)——索氏提取法提取大黄素。
综上,本方法操作简便,灵敏性、准确性、重复性好,可用于香桂化浊胶囊的质量控制。
[1]刘红淼,姜少灏,张振杰,等.正交试验优选香桂化浊胶囊中挥发油的包合工艺 [J].中国药房,2010,21(47):4449.
[2]刘红淼,房桂珍,李艳玲,等.香桂化浊胶囊成型工艺研究[J].中国药房,2011,22(11):990.
[3]刘红淼,杨继章,王云志,等.香桂化浊胶囊质量标准研究[J].中国药房,2011,22(27):2553.
[4]刘红淼,杨继章,李艳玲.肉桂油的研究进展[J].中国药房,2011,22(27):2579.
[5]王 丽,张山川,徐 珽,等.高效液相色谱法测定咽炎颗粒中大黄素的含量[J].中国药房,2007,18(9):684.
[6]宁 洁.HPLC法测定抗眩晕颗粒中大黄素和大黄酚[J].中草药,2010,41(5):749.
