空位缺陷对氧分子在方铅矿(100)表面吸附的影响
2012-11-24蓝丽红陈建华李玉琼
蓝丽红,陈建华,李玉琼,陈 晔,郭 进
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西民族大学 化学化工学院,广西高校化学与生物转化过程新技术重点实验室,南宁 530006;4. 广西大学 物理科学与工程技术学院,南宁 530004)
空位缺陷对氧分子在方铅矿(100)表面吸附的影响
蓝丽红1,3,陈建华2,李玉琼2,陈 晔2,郭 进4
(1. 广西大学 化学化工学院,南宁 530004;2. 广西大学 资源与冶金学院,南宁 530004;3. 广西民族大学 化学化工学院,广西高校化学与生物转化过程新技术重点实验室,南宁 530006;4. 广西大学 物理科学与工程技术学院,南宁 530004)
应用基于密度泛函理论(DFT)的第一性原理,采用广义梯度近似(GGA)和平面波超软赝势方法,研究空位缺陷对氧分子在方铅矿(100)表面吸附行为的影响,并比较和分析它们及理想表面的Mulliken电荷布居、电子密度差图和态密度等。结果表明:铅空位比硫空位难形成,两种空位缺陷表面对氧分子有强烈的化学吸附作用,其吸附能均高于理想表面的,说明空位缺陷可以促进氧分子在方铅矿表面的吸附。氧分子在理想表面及铅空位表面发生了解离吸附,氧原子与硫原子形成了共价键;氧分子在硫空位表面没有发生解离吸附,氧原子与表面的铅原子表现出较强的离子相互作用力。
方铅矿;空位缺陷;氧分子吸附;密度泛函理论
铅是一种主要的金属材料,广泛应用于众多的工业领域,它主要来源于方铅矿的冶炼,泡沫浮选是分离和富集方铅矿最有效的方法。方铅矿的浮选是一个电化学过程,在此过程中氧一直扮演着重要角色。早在20世纪50年代末,国外就有文献报道硫化矿物的表面氧化与其浮选行为之间存在密切的关系[1]。方铅矿具有良好的无捕收剂浮选行为,通过调控氧化程度可使硫化矿物表面形成疏水元素硫或亲水硫酸盐,来实现无捕收剂浮选与分离[2-3]。孙水裕等[4]用伏安曲线和量子化学计算方法研究方铅矿表面的阳极氧化和氧化过程中电子转移的微观机制,结果表明,方铅矿表面荷负电的硫原子能转变成电中性的单质硫,导致方铅矿表面疏水,从而提高其可浮性。用不同方法和手段探讨方铅矿表面与氧分子相互作用机理成为选矿界理论研究的热点[5-9]。
由于成矿环境复杂,自然方铅矿晶体中经常存在各种缺陷,其中晶体结构中阴阳离子空位是一种较为常见的缺陷,导致形成非化学计量的方铅矿晶体结构,并对方铅矿的半导体性质及浮选过程的电化学行为产生显著的影响[10]。陈建华等[11]采用密度泛函理论研究了空位缺陷对方铅矿半导体电子结构及浮选行为的影响,研究结果表明,空位缺陷显著影响方铅矿半导体电子结构,铅空位缺陷使方铅矿费米能级降低,带隙变窄;而硫空位则与铅空位的结果相反,并使得方铅矿由p型半导体变为n型。胡熙庚[12]指出含有阳离子空位的方铅矿有利于黄药的吸附,而含有阴离子空位的方铅矿则不利于黄药的吸附。格列姆博茨基[13]对不同矿床的3种方铅矿的研究表明: 矿物的电子密度及空穴密度比值会影响黄药在其表面的吸附量, 黄药吸附量随空穴密度比值增大而变小,从而使其可浮性变差。
方铅矿表面铅空位和硫空位分别代表方铅矿表面电子富集和缺失两种表面态。另外,药剂在矿物表面的吸附是不均匀的,药剂与矿物的作用首先从活性中心开始,然后往外延伸[10],空位缺陷正好是方铅矿表面的活性中心,因此,研究氧分子在含空位缺陷方铅矿表面的吸附对于了解方铅矿表面性质对其无捕收剂浮选及氧在电化学浮选中的作用具有重要的意义。本文作者通过构造含多层原子的矿物表面层晶模型,采用密度泛函理论对氧分子在方铅矿空位缺陷表面上的吸附方式、吸附能、表面电荷分布和电子转移及表面态进行详细的研究,建立空位缺陷对方铅矿表面氧化的微观模型。
1 计算方法和模型
1.1 计算方法
本研究采用基于密度泛函理论和平面波赝势方法的CASTEP软件[14-15]进行计算。交换关联泛函采用广义梯度近似(GGA)下的PW91梯度修正近似。平面波截断能经过测试后对方铅矿选取280 eV。表面由优化过的体相切出,并在吸附前进行优化,为了消除相邻吸附分子之间的相互影响,采用(4×2)表面层晶模型。对价电子和离子实的相互作用势的描述采用超软赝势(USP)[16],Brillouin区的积分对方铅矿采用 1×2×1的Monkhorst-Pack(MP)k点网络[17-18]。几何优化采用BFGS算法,方铅矿优化的收敛标准如下:能量收敛标准为 2.0×10-5eV/atom,原子位移的收敛标准为0.000 2 nm,原子间作用力的收敛标准为0.5 eV/nm,晶体内应力收敛标准为0.1 GPa;自洽迭代收敛精度为2.0×10-6eV/atom;对于氧分子,则将其放入一个1.5 nm × 1.5 nm × 1.5 nm 的盒子中,计算时采用Gamma点。所有计算都采用自旋极化参数。
1.2 计算模型
方铅矿晶体呈立方体,属立方面心格子,空间对称结构为 Fm3m,分子式为 PbS,属等轴晶系,每个单胞包含4个PbS分子,每个硫原子分别与6个相邻的铅原子配位,而每个铅原子也分别与6个相邻的硫原子配位,形成八面体构造。常见的解理面为沿 Pb—S键断裂的(100)面,表面上的硫原子分别与5个铅原子配位,铅原子分别与 5个硫原子配位。李玉琼等[19]对含不同原子层及真空层厚度的方铅矿表面模型进行了优化测试;结果表明,对方铅矿采用8层原子层,真空层厚度为 15层时能量最稳定,结构最合理;此外,表面顶部3层原子的弛豫略微明显,更低层的原子的弛豫非常小。因此,本研究在吸附过程中固定基底5层原子层,对表面3层原子进行弛豫。(4×2)理想方铅矿(100)表面侧视图及顶视图见图1。采用8层原子模型时其分子式为S64Pb64,建立表面缺陷时,删去一个表面铅原子则产生一个铅空位缺陷(S64Pb63),删去一个表面硫原子则产生一个硫空位缺陷(S63Pb64),构建空位缺陷模型。李玉琼等[19]对氧分子在理想方铅矿表面的吸附位置进行了测试;结果表明,氧分子以平躺于穴位且两个氧原子分别对应着两个硫原子吸附在理想表面时的吸附能最低,吸附最稳定。因此,在空位缺陷表面吸附时将氧分子平躺置于空位缺陷位置,并将两个氧原子对应着缺陷周围两个硫原子进行优化计算。
1.3 能量计算
空位缺陷形成能和氧分子在方铅矿表面的吸附能分别按式(1)和(2)计算。

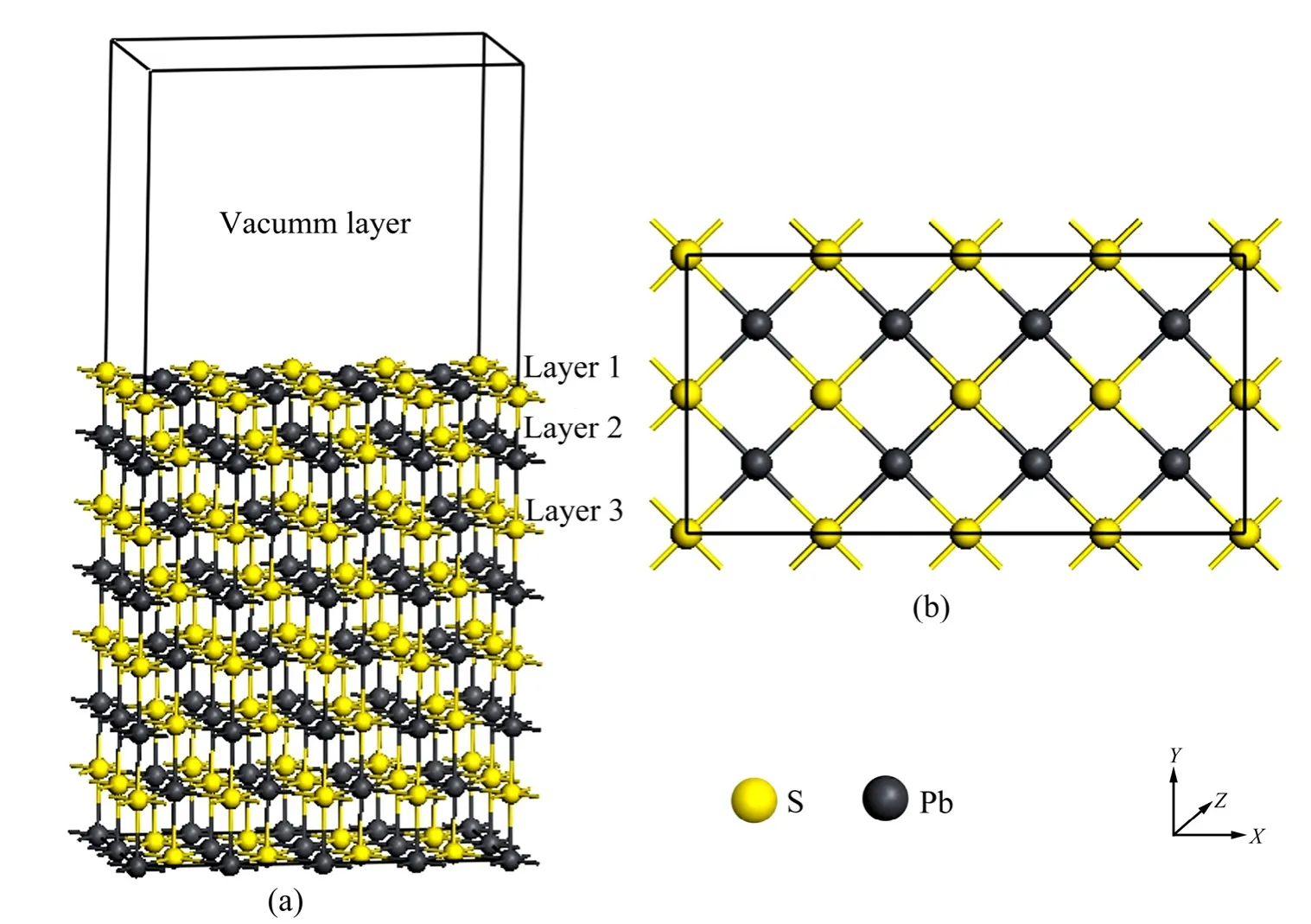
图1 (4×2)方铅矿(100)表面层晶模型Fig. 1 Galena (100) surface (4×2): (a) Side view; (b) Top view (Arrows are indicators of X, Y and Z axes)
式中:Efor为空位缺陷形成能;EX-vacancy为空缺X原子时的总能量(X可指S或Pb原子);Eperfectsurface为理想方铅矿表面总能量;μX为原子X的总能量;Eads为氧分子吸附后的吸附能;EO2/surface为氧分子在表面吸附后体系的总能量;EO2和Esurface分别为吸附前氧分子和方铅矿(可以指理想方铅矿及其空位表面)的总能量。形成能越低,则空位缺陷越易形成,反之亦然;同理,吸附能越低表明吸附越稳定,反之则稳定性降低。
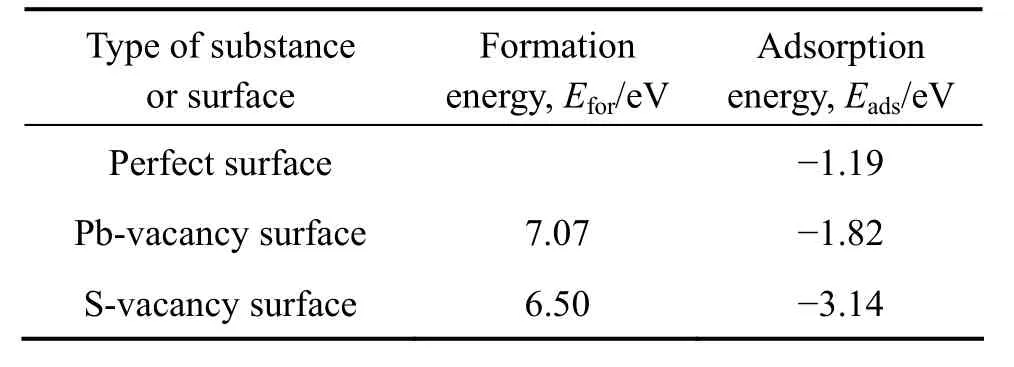
表1 空位缺陷形成能及氧气在不同表面的吸附能Table1 Formation energy of vacancy defect and adsorption energy of O2 on different surfaces
2 结果与讨论
2.1 氧分子在理想方铅矿及空位缺陷表面的吸附构型分析
表1列出了铅和硫空位方铅矿表面的空位缺陷形成能及其对氧分子的平衡吸附能。而氧气在方铅矿表面的吸附构型见图2。
由表1可知,硫和铅空位的形成能都大于零,说明这两种空位缺陷在常温常压下都不能自发形成,但是在成矿时往往会是高温高压的条件下,所以自然界中会在方铅矿表面形成这两种空位缺陷。氧分子的平衡吸附能由大到小顺序为理想表面(-1.19 eV)>铅空位(-1.82 eV)>硫空位(-3.14 eV),氧分子在空位缺陷表面的吸附能都低于在理想表面的吸附能,说明空位缺陷有利于方铅矿表面对氧的吸附,并且硫空位比铅空位更易吸附氧分子而被氧化。
在图 2中,标出的数字为相应两原子之间的键长。由图2可知,氧分子在理想方铅矿和铅空位表面吸附位置与构型相似,吸附后两个氧原子的键长分别为0.269 8和0.305 5 nm,与自由氧分子的键长0.124 1 nm相比较,氧分子都发生了解离,并与表面的硫原子成键。理想表面的氧-硫键长分别为0.164 4和0.164 7 nm,铅空位表面的氧-硫键长分别为0.154 9和0.155 6 nm,表明氧原子与铅空位表面的硫原子之间的键合更为紧密,氧化更强。氧分子在硫空位表面吸附后没有发生解离,氧-氧键长为0.150 9 nm,氧原子只与铅原子键合,成键的铅原子中有表面第一层硫空位周围的3个铅原子(Pb2、Pb3、Pb4)和第二层硫空位对应位置下的一个铅原子(Pb5)。
从键的电荷密度图可以清楚地看到氧原子与表面硫和铅原子之间的成键,如图3所示,白色表示电荷密度为零,另外图中的数字为键的Mulliken布居,布居值越大,表明键的共价性越强,布居值越小表明离子间的作用力越强。从图3(a)和(b)可以看出,在理想方铅矿和铅空位表面氧原子与硫原子之间的电子云重叠较大,表明它们之间形成较强的共价键,而铅空位表面的O—S键的共价性更强,键的布居值也比理想表面的大。而从图3(c)与(d)可知,硫空位表面上的氧原子与其周围的铅原子电子云几乎没有重叠,键的布居值为零或是近似等于零,说明它们之间没有形成共价键,而是离子间存在作用力,形成比较完美的离子键[20]。
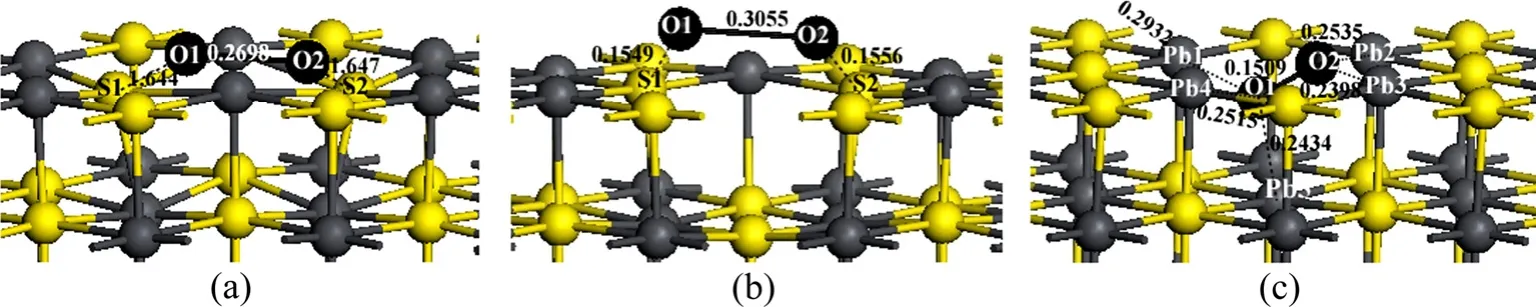
图2 氧分子在理想方铅矿及其空位表面(100)面的平衡吸附构型Fig. 2 Equilibrium adsorption of O2 on different galena (100) surfaces (unit: nm): (a) Perfect surface; (b) Pb-vacancy surface;(c) S-vacancy surface
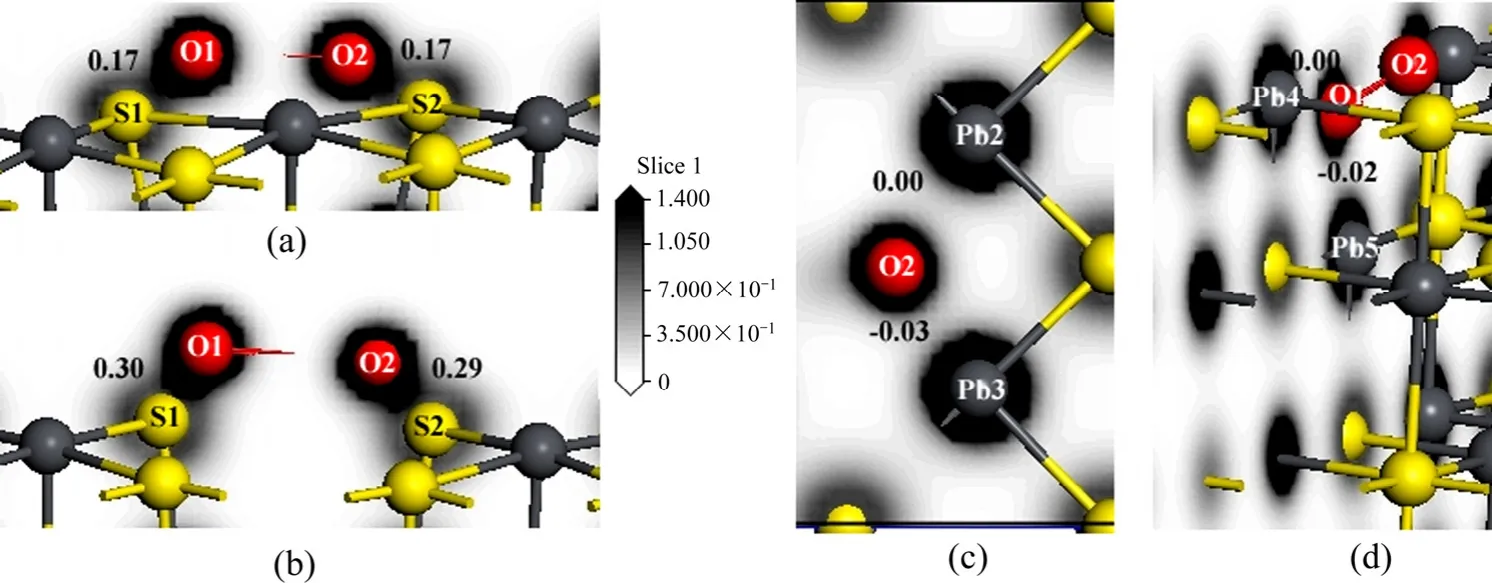
图3 氧分子吸附在方铅矿表面的电荷密度Fig. 3 Electron density maps of galena after O2 adsorption: (a) Electron density of perfect surface; (b) Electron density of Pb-vacancy surface; (c) Top view of electron density of S-vacancy surface; (d) Side view of electron density of S-vacancy surface
2.2 方铅矿表面原子Mulliken电荷分析
图4所示为方铅矿理想和空位表面吸附氧分子前后表面原子的Mulliken电荷,原子上的数字表示电荷值,单位为e,显示图为顶视图。在理想的方铅矿(100)表面上,铅原子带正电荷 0.61e,而硫原子带负电荷-0.68e(见图4(a))。铅空位之后暴露出配位更低、活性更高的硫原子,因此使氧分子的吸附增强。比较图4(e)和(a)可知,硫空位的存在使得其周围的 4个铅原子(Pb1至Pb4)荷电量由+0.61e降至+0.48e,其对周围硫原子的荷电量影响不大,说明硫空位存在使表面铅原子得到电子,从而使表面的正电荷值降低,表面累积的电子增多,氧分子更容易从硫空位表面获得电子,同时硫空位的产生使周围铅原子的配位降低,活性增强,也有利于氧分的吸附。
氧分子吸附后对其周围原子的电荷影响较为明显。由图4(d)中可知,在铅空位表面与氧原子成键的S1和S2原子所带电荷已从原来的负电荷到吸附氧后带正电荷(+0.28e),这比理想表面吸附氧分子后S1和S2原子所带正电荷量(+0.07e)要大,说明在铅空位表面上氧与硫之间的结合更为紧密。氧对距离稍远的硫原子和铅原子电荷影响很小,靠近氧原子的铅原子Pb1失去少量电荷。另外,氧分子吸附对表面硫原子的构型产生了较为明显的影响,与氧成键的S1和S2原子沿着x轴被排斥开来。由图4(f)中可知,氧分子在硫空位表面吸附后,与氧成键的铅原子则失去较多电子,荷更高的正电荷(Pb2和Pb3的荷电量由+0.48e升至+0.69e,Pb4由+0.48e升至+0.71e),而整个表面上的硫原子荷电量变化都不大。

图4 氧分子吸附前后氧原子及表面原子的Mulliken电荷Fig. 4 Mulliken charges of oxygen atom and surface atoms before and after O2 adsorption: (a) Perfect surface before O2 adsorption;(b) Ideal surface after O2 adsorption; (c) Pb-vacancy surface before O2 adsorption; (d) Pb-vacancy after O2 adsorption; (e) S-vacancy surface before O2 adsorption; (f) S-vacancy after O2 adsorption
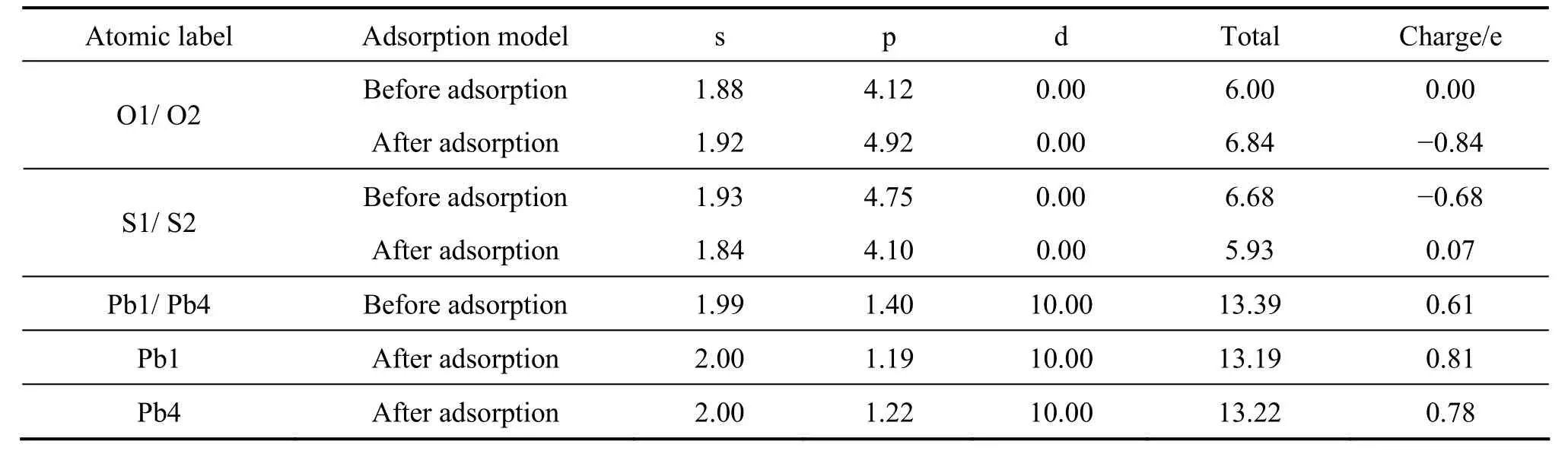
表2 氧分子在理想方铅矿表面吸附前后氧原子及表面原子Mulliken电荷布居Table2 Mulliken charge populations of oxygen atom and surface atom before and after O2 adsorption on perfect galena surface
从更详细的原子电荷布居分析可以清楚地了解原子之间的电荷转移情况,表2~4分别列出了氧分子在理想方铅矿及其空位表面吸附前和吸附后的表面原子及氧原子的电荷布居值。在理想及铅空位方铅矿表面上,氧分子吸附后,氧的2p轨道获得大量电子,与氧成键的硫原子(S2)的3s轨道失去少量电子,而3p轨道则失去大量电子;未参与成键的铅原子主要由 6p轨道失去电子。在硫空位表面上,氧的2p轨道获得大量电子,与氧成键的铅原子(Pb2、 Pb3、 Pb4 和Pb5)主要由 6p轨道失去较多电子,而周围未与氧成键的铅原子(Pb1)的 6p轨道也失去一些电子。由此可知,氧分子与方铅矿表面的反应主要由硫原子3p、铅原子6p以及氧原子的2p轨道共同参与,并且电子由硫和铅p轨道向氧的p轨道转移,此外,铅的d轨道电子没有参与反应。
图5显示了理想表面和空位表面吸附氧分子后的电荷差分密度,黑色区域表示电子富集,白色区域表示电子缺失,背景色表示电子密度为零。由图5可以看到理想、铅空位及硫空位方铅矿3种表面吸附氧原子后,氧原子周围电子富集而与之配位的硫原子及铅原子周围则呈电子缺失状态。
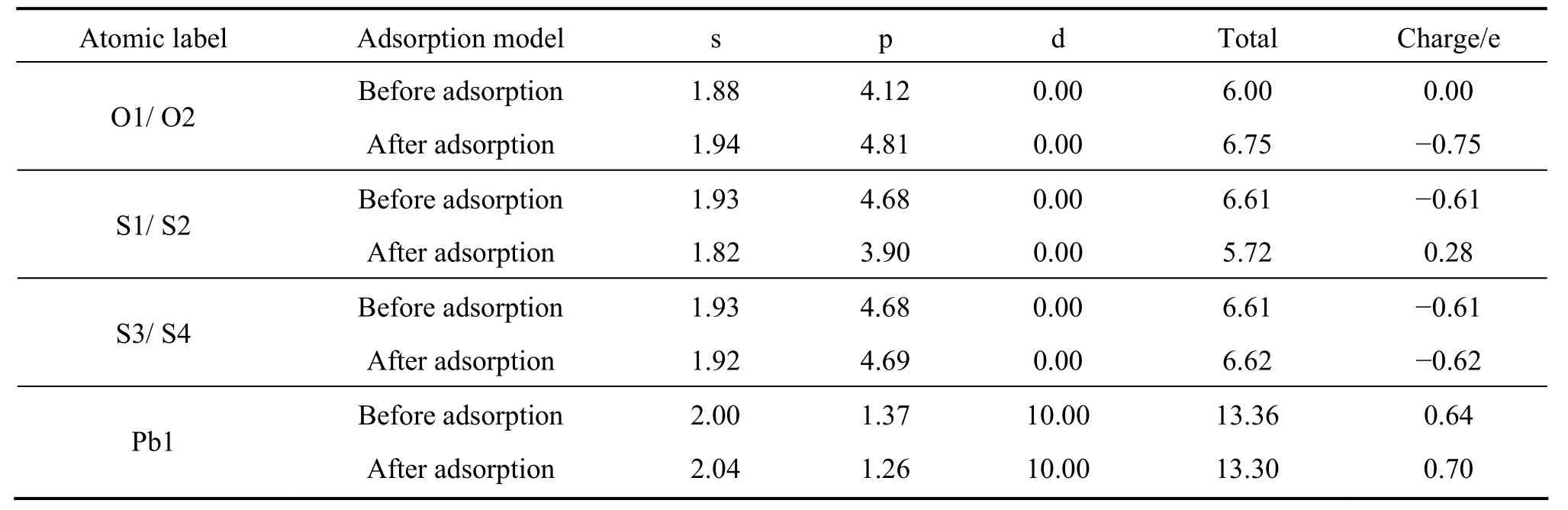
表3 氧分子在铅空位方铅矿表面吸附前后氧原子及表面原子的Mulliken电荷布居Table3 Mulliken charge populations of oxygen atom and surface atom before and after O2 adsorption on galena surface with Pb-vacancy
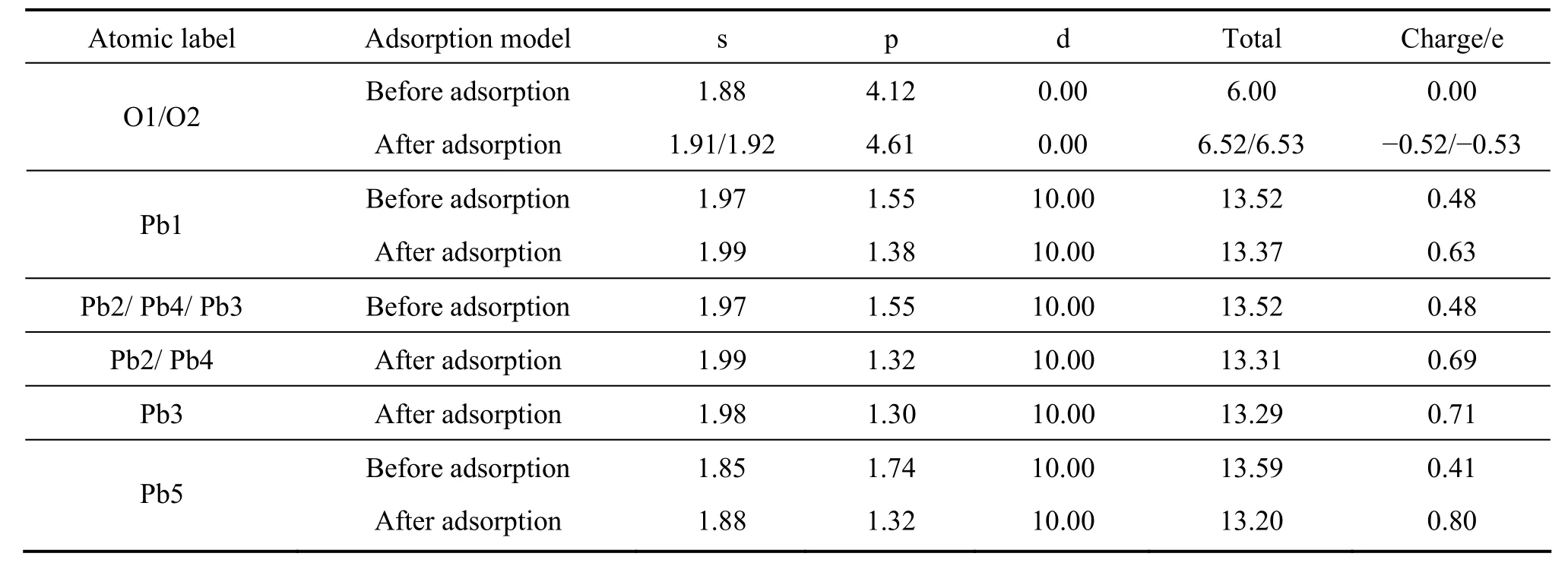
表4 氧分子在硫空位方铅矿表面吸附前后氧原子及表面原子的Mulliken电荷布居Table4 Mulliken charge populations of oxygen atom and surface atom before and after O2 adsorption on galena surface with S-vacancy
2.3 空位缺陷对方铅矿表面态的影响
方铅矿表面吸附氧的能力取决于其表面电子结构,表面空位缺陷的存在改变了空位周围原子的电子结构,从而改变了电子的反应活性。表面原子在费米能级周围的态密度分布越多,其电子的反应活性就越强[21]。图6所示为理想和含空位缺陷方铅矿表面态密度以及表面原子的态密度。由图6可知,空位缺陷的存在使表面态密度发生了显著的变化。与理想方铅矿表面相比较,铅空位表面态密度向高能方向移动,在费米能级附近的态密度分布量明显增大;而硫空位表面态密度向低能量移动,在费米能级处出现一个新的态密度峰。比较空位表面与理想表面上的相同原子态密度变化,发现铅空位表面上的S1原子3p态在费米能级处的分布明显增多,硫原子活性增强,更容易失去 3p轨道上的电子而与氧分子反应;在硫空位表面上,在费米能级处产生了由Pb3原子的6p态贡献的新态密度峰,铅原子活性增强,铅原子6p轨道上的电子更容易失去而与氧分子反应。
以上计算结果表明,空位缺陷的存在使表面铅和硫原子的活性增强,从而有利于浮选药剂分子与方铅矿表面的作用。
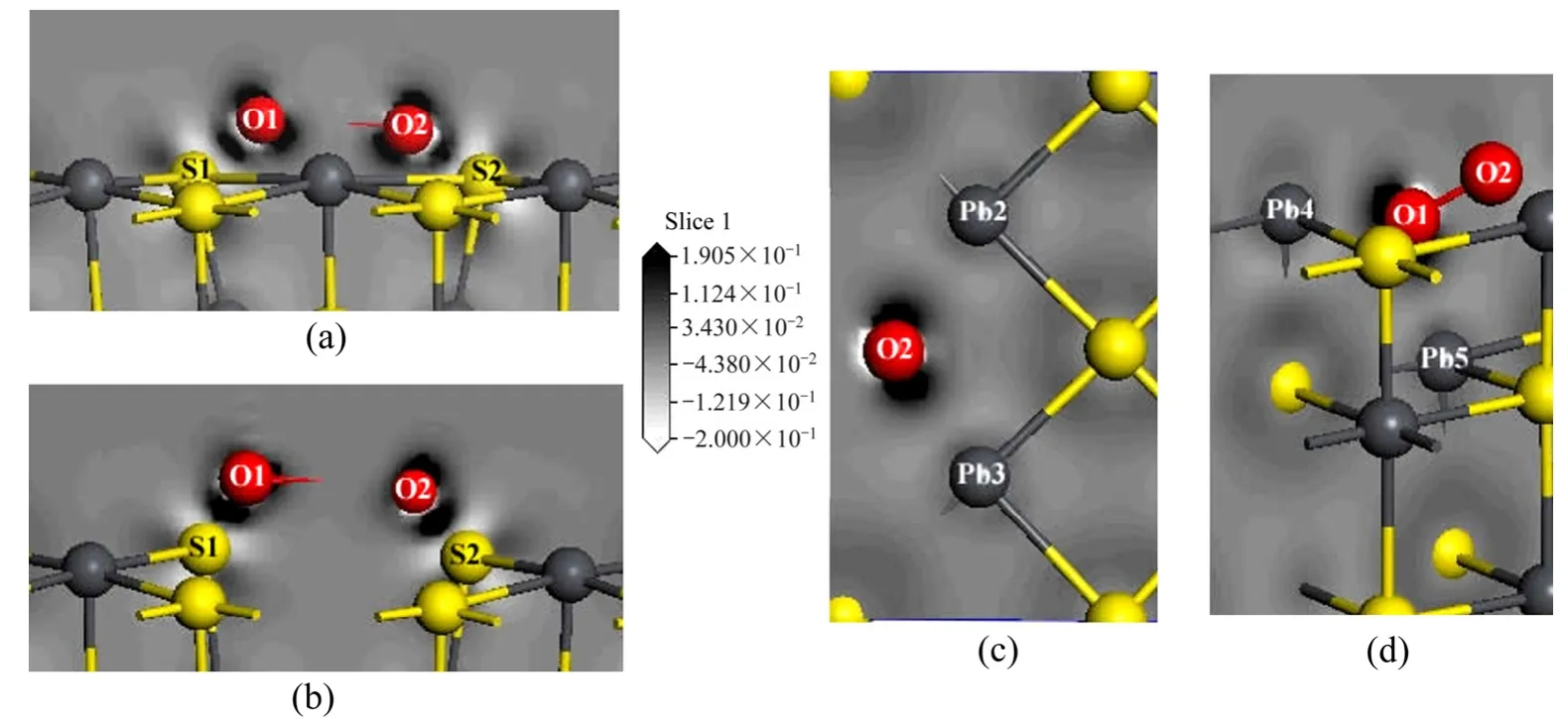
图5 氧分子吸附在方铅矿表面的差分电荷密度Fig. 5 Electron density difference maps of galena after O2 adsorption: (a) Electron density difference of perfect surface;(b) Electron density difference of Pb-vacancy surface; (c) Top view of electron density difference of S-vacancy surface; (d) Side view of electron density difference of S-vacancy surface
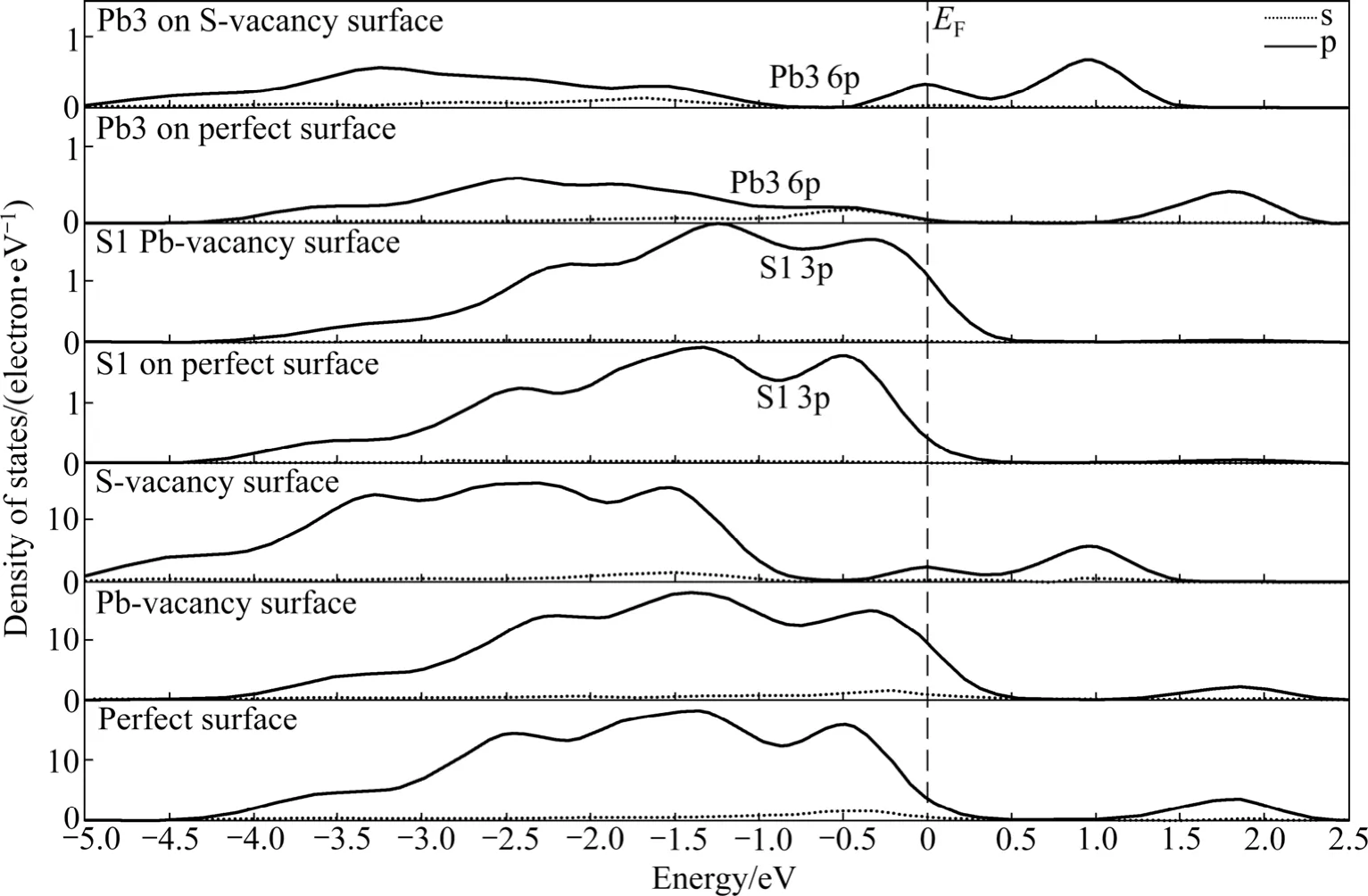
图6 空位缺陷和理想方铅矿表面、表面硫原子及铅原子态密度Fig. 6 Density of states of galena surface, sulfur atom and lead atom on galena surface
2.4 氧分子吸附对方铅矿表面态的影响
从前面的原子电荷布居分析可知,氧分子在方铅矿表面吸附时,主要由氧原子、硫原子和铅原子的 p轨道参与相互作用。图7~9所示分别为氧分子吸附前后理想方铅矿和空位表面态密度及主要参与反应原子的态密度。如图7和8所示,吸附前自由氧分子的态密度图在-6 eV和-4.5 eV处有一个分别由成键σ2pz态和组成的态密度峰;费米能级处的态密度则由半满的反键态组成;约1.8 eV处存在一个空反键态。这与氧分子的外层p电子组态理论排布相一致[22]。在理想及空位缺陷方铅矿表面上吸附后,氧分子及方铅矿表面的态密度均发生了显著的变化。氧分子的态密度整体向低能级移动,氧的2p轨道电子态密度在-6.0~+1.0 eV范围内呈连续分布状态,且主要集中在-5.5~-3.5 eV和-1.0~+0.5 eV两个能量范围内。
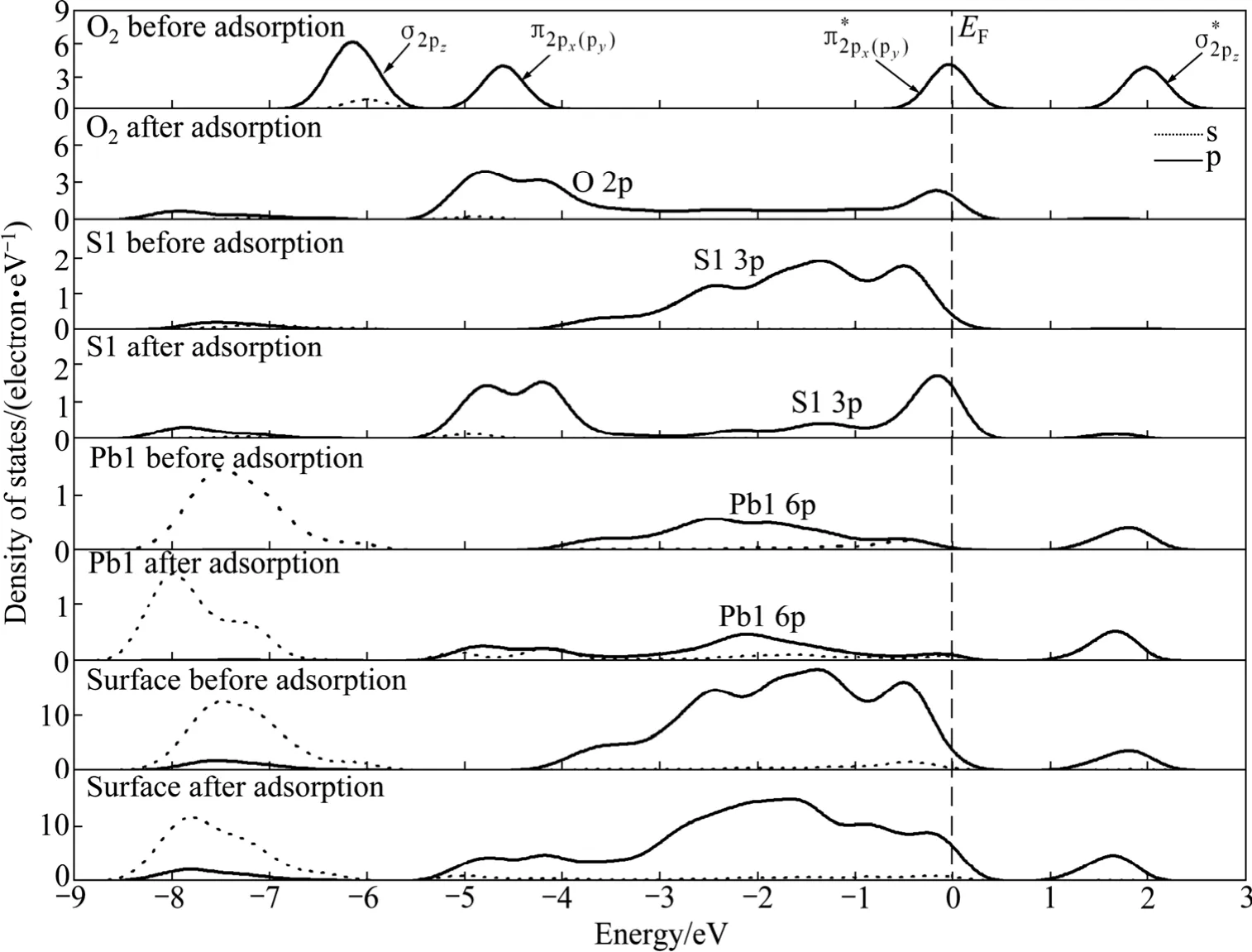
图7 氧分子吸附前后理想方铅矿表面原子和氧分子的态密度Fig. 7 Density of states of surface atom and oxygen before and after O2 adsorption on perfect galena surface
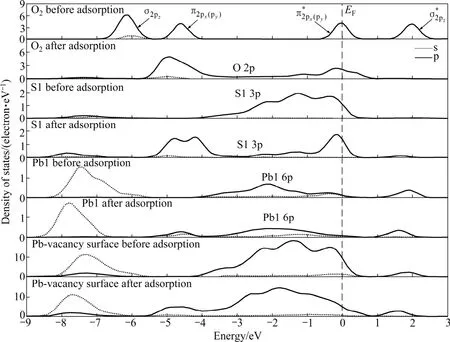
图8 氧分子吸附前后铅空位方铅矿表面原子和氧分子的态密度Fig. 8 Density of states of surface atom and oxygen before and after O2 adsorption on S-vacancy galena surface
在理想及铅空位方铅矿表面上,与氧键合的 S1和S2原子的态密度相同,因此在图7和图8中只显示S1原子的态密度。由图7~8可知,氧分子吸附对理想及铅空位方铅矿表面和表面原子的态密度影响相近。原来处于-4.0~ +0.5 eV范围的连续硫(S1)3p电子态由于氧分子吸附而在-5.0 ~ -4.0 eV和-1.0 ~ 0 eV能量范围内形成两个集中态,并且硫的 3p态与氧的2p态在-0.2 eV能量处发生较强的杂化作用。氧分吸附后对周围铅原子的态密度也产生了一定的影响。Pb1原子的整体态密度向低能方向移动了较小距离且6p电子态密度降低。
在硫空位表面上,氧分子与表面第一层铅原子(Pb2、Pb3、 Pb4)和第二层铅原子(Pb5)发生相互作用。氧分子吸附后Pb2、Pb3、Pb4的态密度变化非常相似,因此在图9中只显示Pb3和Pb5原子的态密度。由图9可以看出,由于氧分子的吸附,表面态密度整体向高能方向移动,Pb3和Pb5原子的态密度明显降低并向高能方向移动,在费米能级处的6p态密度峰消失,此外它们的6p态与氧的2p态在-4.3 eV能量处发生杂化。
由前面的计算结果可知,空位缺陷的存在有利于氧分子的吸附,从而促进黄药在方铅矿表面的电化学作用。
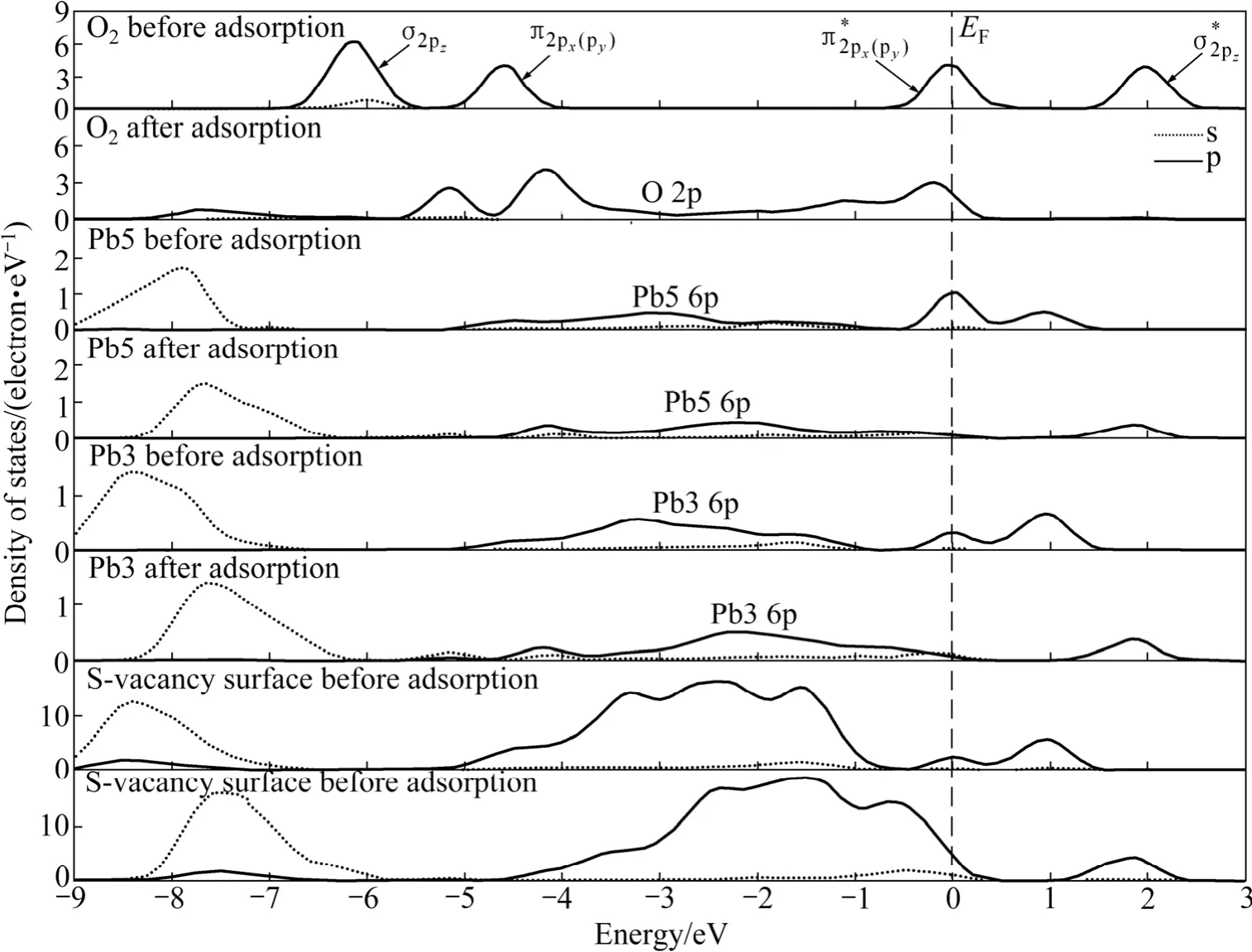
图9 氧分子吸附前后硫空位方铅矿表面原子和氧分子的态密度Fig. 9 Density of states of surface atom and oxygen before and after O2 adsorption on S-vacancy galena surface
3 结论
1) 方铅矿表面吸附氧的能力取决于其表面电子结构,表面空位缺陷的存在改变了空位周围原子的电子结构,从而增强了方铅矿表面吸附活性。其中铅空位使硫原子态密度向费米能级附近移动,增强了硫原子吸附活性,而硫空位则导致了铅原子态密度向费米能级附近移动,增强了铅原子吸附活性。
2) 铅空位和硫空位的存在使得方铅矿的表面周期性遭到破坏,铅空位和硫空位成为氧分子吸附的活性中心,导致氧分子在含空位方铅矿表面的吸附增强。在3种方铅矿表面的吸附能力由大到小顺序是:硫空位>铅空位>理想表面。氧分子在理想及铅空位表面吸附构型相似,均发生了解离吸附,氧原子与表面硫原子形成了共价键,而在硫空位表面没有发生解离吸附,氧原子与表面的铅原子形成了较完美的离子键。
3) 空位缺陷的存在有利于浮选药剂分子在方铅矿表面的作用,而含空位缺陷的方铅矿被氧化后与黄药的电化学作用将增强。
REFERENCES
[1] PLASKIN I N. Interaction of minerals with gases and reagents in flotation [J]. Mining Engineering, 1959, 214: 319-324.
[2] WOODS R. Electrochemical potential controlling flotation [J].International Journal of Mineral Processing, 2003, 72(1/4):151-162.
[3] WOODS R. Recent advances in electrochemistry of sulfide mineral flotation [J]. Transactions of Nonferrous Metals Society of China, 2000, 10(Special Issue): 26-29.
[4] 孙水裕, 王淀佐, 李柏淡. 方铅矿自诱导浮选的电化学和量子化学研究[J]. 有色金属, 1993, 45(2): 32-37.
SUN Shui-yu, WANG Dian-zuo, LI Bo-dan. Electrochemical and quantum-chemical investigations on selfinduced flotation of galena [J]. Nonferrous Metals, 1993, 45(2): 32-37.
[5] 王淀佐, 龙翔云, 孙水裕. 硫化矿的氧化与浮选机理的量子化学研究[J]. 中国有色金属学报, 1991, 1(1): 15-23.
WANG Dian-zuo, LONG Xiang-yun, SUN Shui-yu. Quantum chemical mechanism on the surface oxidation and flotation of sulfide minerals [J]. The Chinese Journal of Nonferrous Metals,1991, 1(1): 15-23.
[6] BECKER U, HCKHELLA M F Jr. The calculation of STM images, STS spectra, and XPS peak shifts for galena: New tools for understanding mineral surface chemistry [J]. Geochimica et Cosmochimica Acta, 1996, 60(13): 2413-2426.
[7] SMART R S C, AMARANTIDIS J, SKINNER W M,PRDSTIDGE C A, LA VANIER L, GRANO S R. Surface analytical studies of oxidation and collector adsorption in sulfide mineral flotation [J]. Scanning Microscopy, 1998, 12(4): 553-583.
[8] GERSON A R, O’DEA A R. A quantum chemical investigation of the oxidation and dissolution mechanisms of galena [J].Geochimica et Cosmochimica Acta, 2003, 67(5): 813-822.
[9] MUSCAT J, GALE J D. First principles studies of the surface of galena PbS [J]. Geochimica et Cosmochimica Acta, 2003, 67(5):799-805.
[10] 胡为柏. 浮选[M]. 北京: 冶金工业出版社, 1983: 27-30.HU Wei-bo. Flotation [M]. Beijing: Metallurgical Industry Press,1983: 27-30.
[11] 陈建华, 王 檑, 陈 晔, 李玉琼, 郭 进. 空位缺陷对方铅矿电子结构及浮选行为影响的密度泛函理论[J]. 中国有色金属学报, 2010, 20(9): 1815-1821.
CHEN Jian-hua, WANG Lei, CHEN Ye, LI Yu-qiong, GUO Jin.Density functional theory of vacancy defects on electronic structure and flotation of galena [J]. The Chinese Journal of Nonferrous Metals, 2010, 20(9): 1815-1821.
[12] 胡熙庚. 有色金属硫化矿选矿[M]. 北京: 冶金工业出版社,1987: 172.
HU Xi-geng. Mineral processing of nonferrous sulphide ores[M]. Beijing: Metallurgical Industry Press, 1987: 172.
[13] B A 格列姆博茨基. 浮选过程物理化学基础[M]. 北京: 冶金工业出版社, 1981: 191.
GLEMBOTSK B A. Foundation of physical chemistry in the process of flotation [M]. Beijing: Metallurgical Industry Press,1981: 191.
[14] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J,PROBERT M I J, REFSON K, PAYNE M C. First principles methods using CASTEP [J]. Zeitschrift fur Kristallographie,2005, 220(5/6): 567-570.
[15] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J,HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[16] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Physical Review B, 1990,41(11): 7892-7895.
[17] MONKHORST H J, PACK J D. Special points for Brillouinzone integrations [J]. Physical Review B, 1976, 13(12): 5188-5192.
[18] PACK J D, MONKHORST H J. Special points for Brilliouinzone zntegrations—A reply [J]. Physical Review B, 1977, 16(4):1748-1749.
[19] 李玉琼, 陈建华, 蓝丽红, 郭 进. 氧分子在黄铁矿和方铅矿表面的吸附[J]. 中国有色金属学报, 2012, 22(4): 1184-1194.
LI Yu-qiong, CHEN Jian-hua, LAN Li-hong, GUO Jin. The adsorption of O2on pyrite and galena surfaces [J]. The Chinese Journal of Nonferrous Metals, 2012, 22(4): 1184-1194.
[20] 任尚元. 有限晶体中的电子态[M]. 北京: 北京大学出版社,2006: 7.
REN Shang-yuan. Electronic state of limited crystal [M]. Beijing:Beijing University Press, 2006: 7.
[21] SEGALL M D, SHAH R, PICKARD C J, PAYNE M C.Population analysis of plane-wave electronic structure calculations of bulk materials [J]. Phys Rev B, 1996, 54: 16317-16320.
[22] 李炳瑞. 结构化学[M]. 北京: 高等教育出版社, 2004: 85-86.
LI Bing-rui. Structural chemistry [M]. Beijing: Higher Education Press, 2004: 85-86.
Effect of vacancy defects on oxygen molecule adsorption on galena surface (100)
LAN Li-hong1,3, CHEN Jian-hua2, LI Yu-qiong2, CHEN Ye2, GUO Jin4
(1. College of Chemistry and Chemical Engineering , Guangxi University, Nanning 530004, China;2. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;3. Key Laboratory of Chemical and Biological Transformation Process of Guangxi Higher Education Institutes,School of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Nanning 530006, China;4. College of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
The effects of vacancy defects on oxygen molecule adsorption on galena surface (100) were studied by applying the general gradient approximation (GGA) and plane-wave ultrasoft pseudopotential method based on the first principle of density functional theory (DFT). The Mulliken charges population, electron density difference maps and density of states of oxygen atom and surface atoms before and after O2adsorption were analyzed. The results show that the formation of Pb-vacancy on the galena surface (100) is more difficult than S-vacancy, and the chemical adsorption of oxygen molecule on both Pb-vacancy and S-vacancy surfaces occurs, and their adsorption energies are more negative than those on the perfect surface, which indicates that vacancy defect can encourage the oxygen molecule adsorption on galena surface. The dissociated adsorption of oxygen molecule occurs on the perfect and Pb-vacancy surfaces, and a covalent bond is formed between oxygen atom and sulfur atom. The oxygen molecule adsorbing on S-vacancy surface is not dissociated. Ionic interaction between oxygen atom and lead atom is very strong.
galena; vacancy defect; oxygen molecule adsorption; density functional theory
TD923
A
1004-0609(2012)09-2626-10
国家自然科学基金资助项目(51164001);广西省自然科学基金资助项目(桂科自0991082)
2011-08-29;
2011-12-10
陈建华,教授,博士;电话:0771-3232200;E-mail: jhchen1971@sina.com
(编辑 何学锋)
