脂质诱导的p62dok高表达增加肝葡萄糖异生
2012-11-06裴亚萍丁佑铭
裴亚萍, 丁佑铭
(1湖北中医药大学医院,湖北 武汉 430065; 2武汉大学人民医院肝胆腔镜外科,湖北 武汉 430060)
1000-4718(2012)09-1676-05
2012-04-05
2012-06-19
△通讯作者 Tel: 027-88041911-82218; E-mail: youmingding@yahoo.com
脂质诱导的p62dok高表达增加肝葡萄糖异生
裴亚萍1, 丁佑铭2△
(1湖北中医药大学医院,湖北 武汉 430065;2武汉大学人民医院肝胆腔镜外科,湖北 武汉 430060)
目的探讨p62dok表达与肝细胞葡萄糖异生间的相互关系。方法利用小鼠高脂饮食模型,观察p62dok表达与肝组织胰岛素信号转导和葡萄糖异生调节蛋白表达间的关系;利用原代培养的小鼠肝细胞,采用基因沉默和过表达等方法,研究p62dok表达量对胰岛素信号通路和葡萄糖异生的影响。采用蛋白免疫印迹法检测蛋白和磷酸化蛋白表达。结果在高脂饮食处理的小鼠肝组织和游离脂肪酸处理的原代培养小鼠肝细胞中, p62dok的表达量显著增加、蛋白激酶B (Akt)和叉头框蛋白O1 (FoxO1)磷酸化程度降低,葡萄糖异生调节蛋白葡萄糖-6-磷酸酶 (G6Pase)和磷酸烯醇丙酮酸羧激酶(PEPCK)的含量增加。沉默p62dok可增加游离脂肪酸处理的原代培养小鼠肝细胞中Akt和FoxO1磷酸化,降低G6Pase和PEPCK的含量。结论高脂可上调肝细胞 p62dok表达,并通过抑制胰岛素信号转导而调节葡萄糖异生。
p62dok; 肝细胞; 葡萄糖异生; 胰岛素信号转导
1990年,Ellis首次在哺乳动物细胞中发现了一个62 kD的蛋白质[1]。1997年,Carpino等[2]和Yamanashi等[3]先后报道该蛋白质存在于胰岛素敏感细胞中,可发生酪氨酸磷酸化,具有“停靠蛋白”(docking protein)的功能,推测其可能参与胰岛素信号系统的调节,并将此蛋白命名为p62dok(Dok1)。其后研究表明,酪氨酸磷酸化的p62dok可募集p120 Ras GTP酶激活蛋白(p120 Ras GTPase-activating protein,p21rasGAP) 和适配器蛋白(adaptor protein)Nck并与其结合,从而参与胰岛素信号系统中Ras/MAPK通路的调节,对细胞的丝裂活动产生影响[4-5]。此外,p62dok是胰岛素受体酪氨酸激酶的直接底物,其受胰岛素刺激而发生在酪氨酸362和398位点上的磷酸化可导致CHO细胞蛋白激酶B(protein kinase B or PKB, Akt)磷酸化的降低[6]。这一研究结果提示,p62dok可能同样参与了对胰岛素信号通路中磷脂酰肌醇3-激酶/蛋白激酶B(phosphotylinosital 3-kinase/ protein kinase B,PI3K/Akt)信号通路的调节,因而也就可能在胰岛素抵抗的发生中具有重要作用。现有研究证实p62dok具有多重作用,参与对肿瘤细胞生长与转移、细胞分化与增殖等多种重要功能的调节[7-9]。
肝细胞胰岛素抵抗是导致肝细胞葡萄糖异生的重要原因之一,在2型糖尿病的发生和发展中起着关键作用[10-11]。为探讨p62dok对肝细胞胰岛素抵抗和肝细胞葡萄糖异生的调节作用,本研究利用高脂饮食诱导小鼠肝组织或游离脂肪酸诱导原代培养肝细胞的胰岛素抵抗,观察p62dok蛋白表达与胰岛素信号转导和葡萄糖异生间的关系;同时利用基因沉默或过度表达的方法探讨p62dok蛋白表达量对培养肝细胞胰岛素信号转导和葡萄糖异生关键调节蛋白表达的影响;旨在阐明p62dok调节肝细胞葡萄糖异生的可能作用与机制。
材 料 和 方 法
1高脂饮食小鼠模型
C57BL/6高脂饮食小鼠模型采用文献方法[12],雄性C57BL/6小鼠从第6周开始喂养,持续16周后进入实验。
2小鼠肝细胞原代培养
原代肝细胞的分离与培养按文献[13]方法进行,简述如下:6~12周龄小鼠肝脏首先用Hanks缓冲液灌注(缓冲液A),接着用含0.05 % 胶原酶(collagenase)和0.8 kU/L胰蛋白酶(trypsin)的Hanks缓冲液(缓冲液B)灌注;在用缓冲液B灌注期间,利用钳子间歇性夹闭下腹腔大静脉(lower abdominal vena cava);然后摘取肝脏、切碎、通过系列尼龙网滤器过滤;收集细胞,利用缓冲液A洗涤3次,然后悬浮于肝细胞恢复培养液中[DMEM:含10 mg/L葡萄糖、10%胎牛血清、1 μg/L两性霉素B (amphotericin B)、10 kU/L青霉素、50 g/L链霉素、10 g/L庆大霉素、1 nmol/L地塞米松、1 nmol/L胰岛素];细胞培养条件为37 ℃、95% O2和5% CO2。
3试剂、抗体和质粒
p62dokshRNA慢病毒质粒和各种抗体均购自Santa Cruz,其p62dokshRNA的发夹序列为CCGGGCCTACCGATAACCCACCTAACTCGAGTTAGGTGGGTTA-TCGGTAGGCTTTTT。原钒酸钠(sodium vanadate)、抑酶肽(aprotinin)、亮抑蛋白酶肽(leupeptin)等均购自Sigma。
4质粒构建与细胞病毒感染
携带人类全长p62dok的重组慢病毒质粒将按本实验室常规方法构建[14]。病毒制备的方法简述如下:将病毒质粒转入HEK 293FT细胞中,经细胞扩增后,收集上清和细胞,经3次反复冻溶裂解细胞后,离心纯化病毒,测定滴度后-80 ℃保存。病毒滴度的测定采用Cell BioLabs公司的慢病毒快速定量试剂盒(QuickTiterTMLentivirus Quantitation Kit)完成,具体操作按说明书进行。细胞病毒感染方法如下:培养肝细胞与携带p62dok或p62dokshRNA,及其各自相应对照(scrambled)的重组慢病毒共同培养16 h(MOI为20),然后更换培养液,分组实验。
5蛋白免疫印迹
实验结束后,立即取待研究组织或细胞,用4 ℃预冷的PBS(0.01 mol/L, pH 7.4)清除血液或洗涤细胞,然后加入细胞裂解液制备组织或细胞匀浆。细胞裂解液组成如下:50 mmol/L HEPES (pH 7.6)、150 mmol/L NaCl、1% Triton X-100、1 mmol/L PMSF、1 mmol/L sodium vanadate、10 mg/L aprotinin和 10 mg/L leupeptin。组织或细胞溶解液15 000 r/min、4 ℃离心15 min,取上清液冰浴20 min。Bradford法定量组织或细胞裂解液中蛋白浓度,并调整各匀浆蛋白浓度一致。然后,将组织或细胞裂解液(蛋白浓度为2 g/L)与等体积的2× SDS上样缓冲液混合,95 ℃水浴 5 min,离心取上清行SDS/PAGE电泳。电泳条件: 堆积胶恒流10 mA,分离胶恒流15 mA。电转移在Bio-Rad电转移槽装置上进行,电转移条件为180 V、1 h。转移后的硝酸纤维素(nitrocellulose,NC)膜行丽春红可逆染色以确定标准分子蛋白位置和转移效果。免疫印迹按如下方法进行:NC膜装入杂交袋,加封阻液(含1% BSA的TTBS),室温振摇1 h;去除封阻液,加入Ⅰ抗(封阻液1∶1 000稀释抗体),封袋,室温振摇1 h或4 ℃过夜;除去Ⅰ抗,以TTBS洗膜10 min × 4,加入Ⅱ抗,封袋,室温振摇1 h;去除Ⅱ抗,TTBS洗膜10 min × 4,PBS洗膜5 min;加入BCIP/NBT底物显迹液显色3~5 min,蒸馏水洗膜终止反应,晾干后拍照。蛋白免疫印迹至少重复3次以上。
6统计学处理
结 果
1高脂饮食增加肝组织p62dok表达和葡萄糖异生
如图1A所示,高脂饮食可显著增加肝组织p62dok蛋白表达(与正常饮食组比较,P<0.01)。同时,肝组织中Akt T308/S473磷酸化,以及Akt下游叉头框蛋白O1(forkhead box O1 protein,FoxO1)S253磷酸化明显降低,见图1B,表明高脂饮食导致肝组织胰岛素抵抗。为研究肝细胞葡萄糖异生的变化,我们检测了葡萄糖异生调节蛋白葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)和磷酸烯醇丙酮酸羧激酶(phosphoenolpyruvate carboxykinase,PEPCK)的表达,结果发现这些重要调节蛋白表达量明显升高,见图1C;与正常饮食组比较,分别P<0.01和P<0.05,提示高脂饮食可增加肝细胞葡萄糖异生。
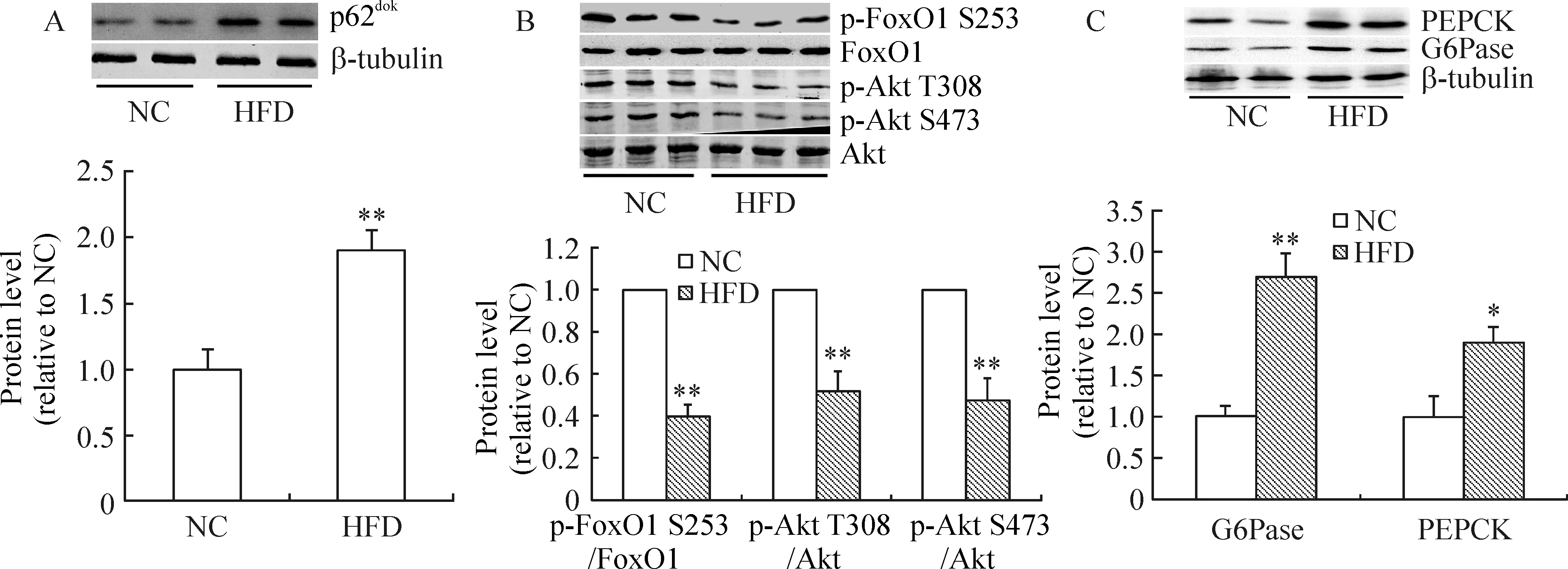

图1高脂饮食对肝组织葡萄糖异生的影响
2游离脂肪酸增加肝细胞p62dok表达和葡萄糖异生
为进一步证实高脂对肝细胞p62dok表达和葡萄糖异生的影响,原代培养的小鼠肝细胞以无血清处理6 h后,予以0.5 mmol/L的棕榈酸盐和(或)100 nmol/L胰岛素处理18 h,结果发现,棕榈酸盐可增加肝细胞p62dok的表达(P<0.05),见图2A,并抑制胰岛素诱导的Akt T308磷酸化和FoxO1 S253磷酸化(P<0.01),见图2B;同时,棕榈酸盐处理可以明显增加胰岛素处理时的G6Pase和PEPCK表达(P<0.01),见图2C。
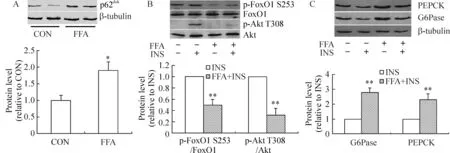
图2游离脂肪酸对原代培养肝细胞葡萄糖异生的影响
3p62dok基因沉默抑制肝细胞葡萄糖异生
p62dok的过度表达可抑制胰岛素诱导的Akt T308磷酸化和FoxO1 S253磷酸化,增加G6Pase和PEPCK的表达,见图3A;而p62dok的沉默可增加胰岛素诱导的Akt T308磷酸化和FoxO1 S253磷酸化,抑制G6Pase和PEPCK的表达,见图3B。上述结果提示p62dok的蛋白表达量与胰岛素抵抗和肝细胞葡萄糖异生呈正相关。
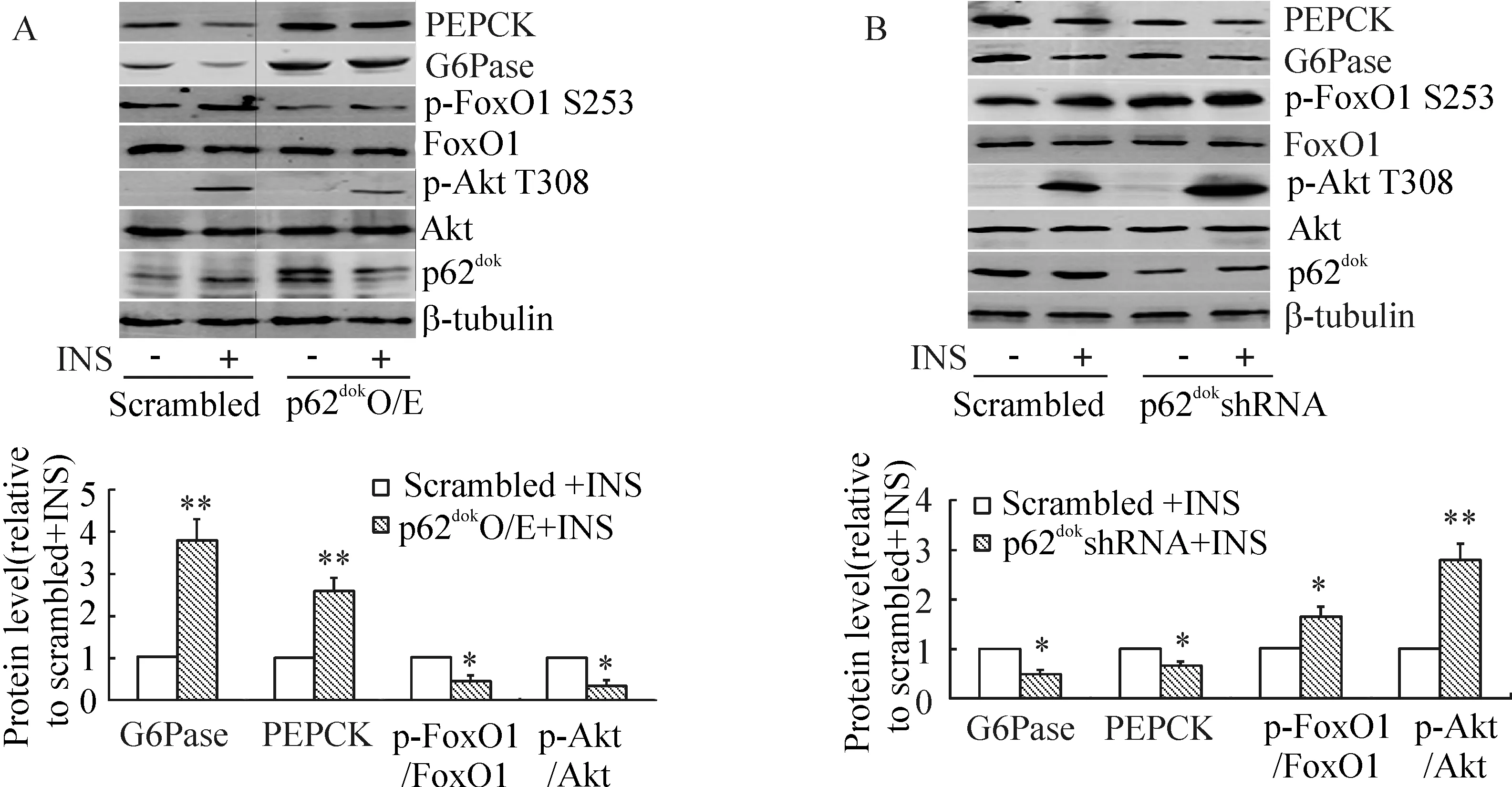
图3p62dok蛋白含量对原代培养肝细胞葡萄糖异生的影响
讨 论
本实验结果发现,高脂饮食或游离脂肪酸处理可增加p62dok表达,导致胰岛素抵抗,增加葡萄糖异生;过度表达p62dok可产生与高脂饮食或游离脂肪酸处理类似的结果;而敲除p62dok可提高胰岛素敏感性,降低肝细胞葡萄糖异生调节蛋白G6Pase和PEPCK的表达。这一结果提示,p62dok高表达所致的胰岛素抵抗是导致脂毒性诱导肝细胞葡萄糖异生增加的重要原因之一。
高脂所导致的脂毒性在肥胖、胰岛素抵抗、2型糖尿病及其并发症的发生发展中具有重要意义,而胰岛素抵抗在其中起到关键作用,其发生与胰岛素信号转导系统PI3K/Akt通路的缺陷密切相关[15]。一般认为,酪氨酸磷酸化的p62dok是一个“停靠蛋白”,对Ras/MAPK信号通路具有抑制作用[1-8]。尽管研究表明,CHO细胞中p62dok磷酸化位点突变可影响胰岛素诱导的Akt磷酸化,但目前还不清楚p62dok是否对其它胰岛素敏感细胞的PI3K/Akt通路产生影响。Hosooka等[12]发现,高脂饮食可导致小鼠脂肪组织p62dok的高表达,敲除小鼠p62dok可有效调节小鼠的能量代谢,提示p62dok在能量代谢中的重要作用。与此相似,我们的结果发现高脂或游离脂肪酸处理可增加肝细胞p62dok表达,且p62dok高表达与胰岛素信号系统PI3K/Akt通路的抑制相一致,表明p62dok有可能参与了对该系统的调节。进一步的研究证实,当肝细胞过度表达p62dok时,Akt磷酸化和FoxO1磷酸化被有效抑制;而沉默p62dok可增加Akt和FoxO1的磷酸化;这些结果充分证实p62dok对肝细胞胰岛素PI3K/Akt信号通路具有抑制作用。
肝细胞胰岛素抵抗是导致肝细胞葡萄糖异生的重要原因之一[10]。正常情况下,胰岛素与其细胞膜的受体结合,并引发受体磷酸化和下游蛋白的磷酸化,通过IRS1/PI3K/PDK1信号转导而激活Akt,包括使Akt T308和Akt S473位点发生磷酸化。激活的Akt导致FoxO1 S253磷酸化,而磷酸化的FoxO1将位于细胞浆内,从而降低葡萄糖异生调节因子,如过氧化物酶体增殖激活受体γ共激活因子1α(peroxisome proliferator-activated receptor gamma coactivator 1 alpha,PGC1α)、G6Pase、PEPCK等的转录,抑制葡萄糖异生过程[11]。在本研究中,高脂诱导的p62dok高表达导致了肝细胞的胰岛素抵抗,Akt活性和FoxO1磷酸化降低,FoxO1将从胞浆转移到胞核内,从而启动G6Pase和PEPCK等基因转录,增加G6Pase和PEPCK蛋白含量,激活葡萄糖异生过程;而敲除p62dok基因则可逆转这一变化。因此,p62dok通过对胰岛素抵抗的调节而控制肝细胞葡萄糖的异生活动。
总之,本研究结果表明:高脂可诱导肝细胞p62dok高表达,并通过p62dok抑制胰岛素PI3K/Akt/FoxO1信号通路,从而增加葡萄糖异生。但对高脂如何诱导p62dok增加,以及p62dok通过何种机制抑制Akt活性的等关键问题仍有待进一步阐明。
[1] Ellis C, Moran M, McCormick F, et al. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases[J]. Nature, 1990,343(6256): 377-381.
[2] Carpino N, Wisniewski D, Strife A, et al. p62dok: a constitutively tyrosine-phosphorylated, GAP-associated protein in chroic myelogenous leukemia progenitor cells[J]. Cell,1997,88(1): 197-204.
[3] Yamanashi Y,Baltimore D. Identification of the Abl-and ras GAP-associated 62 KDa protein as a docking protein, Dok[J]. Cell, 1997,88(1):205-211.
[4] Noguchi T, Matozaki T, Inagaki K, et al. Tyrosine phosphorylation of p62Dokinduced by cell adhesion and insulin: possible role in cell migration[J]. EMBO J, 1999,18(7):1748-1760.
[5] Hosooka T, Noguchi T, Nagai H, et al. Inhibition of the motility and growth of B16F10 mouse melanoma cells by dominant negative mutants of Dok-1[J]. Mol Cell Biol, 2001,21(16):5437-5446.
[6] Wick MJ, Dong LQ, Hu D, et al. Insulin receptor-mediated p62doktyrosine phosphorylation at residues 362 and 398 plays distinct roles for binding GTPase-activating protein and Nck and is essential for inhibiting insulin-stimulated activation of Ras and Akt[J]. J Biol Chem, 2001,276(46):42843-42850.
[7] Kawamata A, Inoue A, Miyajima D, et al. Dok-1 and Dok-2 deficiency induces osteopenia via activation of osteoclasts[J]. J Cell Physiol, 2011,226(12):3087-3093.
[8] Janas JA, Van Aelst L. Oncogenic tyrosine kinases target Dok-1 for ubiquitin-mediated proteasomal degradation to promote cell transformation[J]. Mol Cell Biol, 2011,31(13):2552-2565.
[9] Mashima R, Honda K, Yang Y, et al. Mice lacking Dok-1, Dok-2, and Dok-3 succumb to aggressive histiocytic sarcoma[J]. Lab Invest,2010,90(9):1357-1364.
[10]Quinn PG, Yeagley D. Insulin regulation of PEPCK gene expression: a model for rapid and reversible modulation[J]. Curr Drug Targets Immune Endocr Metabol Disord, 2005,5(4):423-437.
[11]Shah P, Basu A, Rizza R. Fat-induced liver insulin resistance[J]. Curr Diab Rep, 2003,3(3):214-218.
[12]Hosooka T, Noguchi T, Kotani K, et al. Dok1 mediates high-fat diet-induced adipocyte hypertrophy and obesity through modulation of PPAR-γ phosphorylation[J]. Nat Med, 2008,14(2):188-193.
[13]Li WC, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes[J]. Methods Mol Biol, 2010,633(1):185-196.
[14]Wang X, Yu W, Nawaz A, et al. Palmitate induced insulin resistance by PKCtheta-dependent activation of mTOR/S6K pathway in C2C12 myotubes[J]. Exp Clin Endocrinol Diabetes, 2010,118(9):657-661.
[15]Boden G. Obesity, insulin resistance and free fatty acids[J]. Curr Opin Endocrinol Diabetes Obes, 2011,18(2):139-143.
Lipid-inducedp62dokup-regulationenhanceshepaticgluconeogenesis
PEI Ya-ping1, DING You-ming2
(1UniversityHospital,HubeiUniversityofChineseMedicine,Wuhan430065,China;2DepartmentofHepatobiliary&LaparoscopicSurgery,People’sHospitalofWuhanUniversity,Wuhan430060,China.E-mail:youmingding@yahoo.com)
AIM: To investigate the potential role of p62dokin the regulation of hepatic gluconeogenesis.METHODSThe expression of p62dok, insulin signaling transduction, and hepatic gluconeogenesis were investigated in the liver tissues of mice treated with high-fat diet (HFD) and in cultured mouse hepatocytes treated with free fatty acid (FFA). The experiments of gene silencing and overexpression were conducted to observe the effects of p62dokon insulin signal transduction and hepatic gluconeogenesis in cultured mouse hepatocytes. Western blotting was used to detect the protein levels and the phosphorylation statue.RESULTSThe increased p62doklevels were found in the liver tissues from HFD-treated mice and FFA-treated hepatocytes. Meanwhile, phosphorylation of Akt and forkhead box O1 protein (FoxO1) was were decreased and the expression of glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase(PEPCK) was increased. Silencing ofp62dokin cultured hepatocytes treated with FFA induced the increase in phosphorylation of Akt and FoxO1, and decrease in the protein levels of G6Pase and PEPCK.CONCLUSIONUp-regulation of p62dokinduced by HFD or FFA enhances hepatic gluconeogenesis via inhibiting insulin signal transduction.
p62dok; Hepatocytes; Gluconeogenesis; Insulin signal transduction
R363
A
10.3969/j.issn.1000-4718.2012.09.024
