高效液相色谱法测定止痢颗粒中黄芩苷含量
2012-11-06郭立恒朱山寅
郭立恒,朱山寅
(1.浙江省桐乡市第二人民医院,浙江 嘉兴 314511; 2.浙江省嘉兴市食品药品检验所,浙江 嘉兴 314001)
止痢颗粒是由黄芩、葛根、黄连、木香、槟榔、地锦草等6味中药组成的复方制剂,具有清热解毒、理气止痢功效,用于急性痢疾,收载于《卫生部药品标准·中药成方制剂(第五册)》,原标准无含量测定项目[1],也尚未见文献报道。黄芩主含黄芩苷,是本方的有效消炎成分之一。为有效控制止痢颗粒的质量,笔者建立了测定该制剂中黄芩苷含量的高效液相色谱(HPLC)法。现报道如下。
1 仪器与试药
高效液相色谱仪(Waters 510/484/745B色谱系统);UV-265型紫外分光光度计(日本岛津)。黄芩苷对照品(中国药品生物制品检定所,供含量测定用,批号为0751-200912);止痢颗粒(市售品,批号分别为 090408,20090513,091225);甲醇为色谱纯,其余试剂均为分析纯;水为重蒸馏水。
2 方法和结果
2.1 色谱条件
色谱柱:Hypersil C18分析柱(150 mm×4.6 mm,5μm);流动相:甲醇-水-磷酸(47∶53∶0.5);流速:1.0 mL/min;测定波长:278 nm;柱温:室温。
2.2 溶液制备
精密称取经60℃减压干燥至恒重的黄芩苷对照品适量,加甲醇制成每1 mL中含0.055 mg的溶液,作为对照品溶液。取样品10 g,研细,精密称取1 g,置25 mL量瓶中,加甲醇约22 mL,超声处理30 min,放置室温,稀释至刻度,摇匀,滤过,取续滤液用0.45μm的微孔滤膜滤过,即得供试品溶液。按处方量制备不含黄芩药材的阴性样品,按供试品溶液制备方法制成阴性对照品溶液。
2.3 方法学考察
干扰试验:照上述色谱条件,吸取对照品溶液、供试品溶液、阴性对照品溶液各20μL,分别注入色谱仪。供试品溶液色谱图中,在与对照品溶液色谱图相应位置上有一相同的色谱峰,而阴性对照品溶液在此保留时间无干扰,见图1。
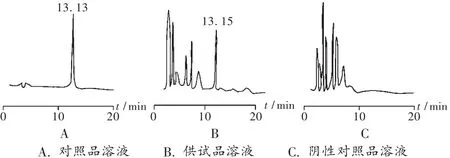
图1 止痢颗粒高效液相色谱图
线性关系考察:精密吸取黄芩苷对照品溶液(0.055 g/L)1.0,1.5,2.0,2.5,3.0,3.5,4.0,4.5,5.0 mL,置 10 mL 量瓶中,用甲醇稀释至刻度,摇匀,配成系列质量浓度的对照品溶液,按上述色谱条件,精密进样20μL。以黄芩苷峰面积积分值 Y为纵坐标、进样量 X(μg)为横坐标绘制标准曲线,得回归方程 Y=7.237×106X+9.973×104,r=0.999 4(n=9)。结果表明,黄芩苷进样量在0.10~0.51μg范围内与峰面积积分值呈良好线性关系。
精密度试验:准确吸取对照品溶液20.0μL,重复进样5次。结果黄芩苷峰面积的 RSD为1.09%(n=5),表明仪器精密度符合要求。
稳定性试验:取同一供试品溶液,分别在制备后 0,2,4,6,8,10 h时进样。结果黄芩苷峰面积的 RSD=1.37%(n=6),表明供试品溶液在10 h内稳定。
重现性试验:取同一批号样品5份,依法制备供试品溶液并测定。结果的 RSD=1.44%(n=6),表明方法重现性良好。
加样回收试验:称取已知含量样品(批号为090408)6份,精密称定,精密加入黄芩苷对照品适量,照2.2项下供试品溶液制备方法制备溶液并按拟订的色谱条件进行测定。结果见表1。
2.4 样品含量测定
分别精密称取不同批号的样品粉末约1 g,依法制备供试品溶液,分别精密吸取20μL,按拟订的色谱条件测定。3批样品中黄芩苷的含量分别为0.453 2,0.449 3,0.450 2 mg/g(n=5)。

表1 黄芩苷加样回收试验结果(n=6)
3 讨论
取对照品溶液,用紫外分光光度计在200~300 nm波长范围内扫描,结果在278 nm波长处有最大吸收,故以此作为测定波长。
曾以乙醇为溶剂进行测定,样品主峰分离不佳,常有列峰现象,对照品峰形同样较差。以乙醇、50%甲醇、甲醇为溶剂对比考察线性关系,黄芩苷进样量相同的情况下,测得的峰面积乙醇最小,甲醇最大,50%甲醇略小于甲醇,r分别为0.948 4,0.980 7,0.999 4(n=6),测得样品中黄芩苷含量的重现性甲醇最好。这说明黄芩苷在乙醇中的溶解度较差,使峰面积产生误差。
比较了几种不同比例流动相的分离效果,要使样品中的黄芩苷与杂质的分离达到要求,且峰形对称而尖锐,必须增加水和磷酸的比例,同时样品的稀释时应改为甲醇。拟订的色谱条件下,黄芩苷与其他组分能达到基线分离,理论板数大于1 500。
[1]WS3-B-0904-91,卫生部药品标准·中药成方制剂(第五册)[S].
