柱前衍生化高效液相色谱法测定莲子心中去甲乌药碱含量
2012-10-25田海峰衣涛金东日
田海峰, 衣涛, 金东日*
(1.延边大学理学院 化学系,吉林 延吉133002;2.吉林烟草工业有限责任公司,吉林 延吉133001)
柱前衍生化高效液相色谱法测定莲子心中去甲乌药碱含量
田海峰1, 衣涛2, 金东日1*
(1.延边大学理学院 化学系,吉林 延吉133002;2.吉林烟草工业有限责任公司,吉林 延吉133001)
以9-芴甲基-N-琥珀酰亚胺基碳酸酯(Fmoc-OSu)为荧光衍生化试剂,建立了柱前荧光衍生化反相高效液相色谱法分析去甲乌药碱的高灵敏分析方法.在硼酸缓冲溶液(p H=8.5)中,去甲乌药碱与Fmoc-OSu在温和的反应条件下反应生成具有荧光性的去甲乌药碱衍生物.采用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色谱柱,用乙腈-水(体积分数比为85∶15)等溶液强度洗脱,以λ=265 nm为激发波长,λ=315 nm为发射波长,对去甲乌药碱衍生物进行了检测.实验结果显示:去甲乌药碱在0.05~20μg/m L范围内呈良好的线性关系(r=0.999 8),其最低检出限(S/N=3)为5.0 ng/m L,加标回收率为94.1%~105.9%,RSD为2.74%(n=5).本方法灵敏度高、选择性好、准确度高,能够满足含去甲乌药碱中药材的质量控制.
去甲乌药碱;柱前荧光衍生化;HPLC;Fmoc-OSu;莲子心
去甲乌药碱(higenamine,HG)是乌头、附子、莲子心等中药中所含有的强心的有效成分.药理实验表明,去甲乌药碱对心血管系统有正力性和正时性效应[1],其特性类似于多巴胺丁胺,有望成为一种新的临床心肌负荷药物.近年来研究发现,去甲乌药碱对iNOS m RNA的表达、脂多糖LPS诱导的NO产物以及对血小板聚集和血栓形成都具有抑制作用,而且对动物实验性DIC也具有改善作用[2].目前,去甲乌药碱的常用测定方法有HPLC-UV[3]、HPLC-ECD[4]、HPLC-ESI-MS[5]、HPLC-FLD[6]等,其中HPLC-UV方法检测灵敏度较低,不适于含微量去甲乌药碱试样的分析;HPLC-ECD和HPLC-ESI-MS方法检测灵敏度虽然较高,但由于ECD和MS检测器较为昂贵,因此限制了其广泛的应用.本文研究者曾用手性荧光衍生化试剂建立了分离去甲乌药碱对映体的HPLC-FLD[7]方法.本文以9-芴甲基-N-琥珀酰亚胺基碳酸酯(Fmoc-OSu)为荧光衍生化试剂,用HPLC-FLD建立了去甲乌药碱的测定方法,并用此方法测定了莲子心中去甲乌药碱的含量.
1 实验部分
1.1 仪器与试剂
仪器有Shimadzu LC-10AVP高效液相色谱仪(日本岛津公司),RF-10AXL荧光检测器,Class-VP色谱工作站,AS20500A超声波清洗器(天津奥特赛恩斯仪器有限公司),HB-100恒温金属浴(杭州大和热电子有限公司).
去甲乌药碱(纯度大于97%)由江西中药研究所提供,Fmoc-OSu(纯度大于98%)购于J&K CHEMICAL LTD,乙腈、甲醇为色谱纯(山东禹王实业有限公司),氢氧化钾、氯化钾为优级纯,硼酸为分析纯试剂.
配制1mg/m L的去甲乌药碱和5 mmol/L的Fmoc-OSu,于冰箱中保存,实验前稀释到所需浓度;用0.2 mol/L氢氧化钾溶液调节硼酸缓冲溶液p H值至8.5.
1.2 衍生化反应
分别取一定浓度的去甲乌药碱标准溶液、Fmoc-OSu乙腈溶液(待测物浓度的125倍)和硼酸缓冲溶液(p H=8.5)各10μL混合于聚丙烯管中,用涡旋搅拌器搅拌1 min,在室温下反应50 min.
1.3 样品前处理
将莲子心研磨成粉末,精确称取莲子心粉末2.50 g放入100 m L锥形瓶中,加入25 m L甲醇,在超声波清洗器中超声30 min(50℃下),过滤.上述操作重复进行3次,合并滤液.将滤液在45℃下减压蒸馏浓缩旋至近干,用乙腈-水(体积分数比为1∶1)定容至25 m L.经0.45μm微孔滤膜过滤,取1 m L滤液用水定容至200 m L.
1.4 色谱条件
采 用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色谱柱,流动相为乙腈-水(体积分数比为85∶15),柱温为室温,进样量为10μL;荧光检测波长为λex=265 nm,λem=315 nm.
2 结果与讨论
2.1 衍生化反应条件
Fmoc-OSu作为一种柱前衍生化试剂,具有过量的衍生化试剂不干扰分离、其衍生物稳定性较好、基体干扰不明显、衍生选择性较好等[7]特点,已用于氨基酸、多肽的分离分析.因此,本研究以Fmoc-OSu作为分离分析去甲乌药碱的衍生化试剂.图1为去甲乌药碱与Fmoc-OSu的衍生化反应式.
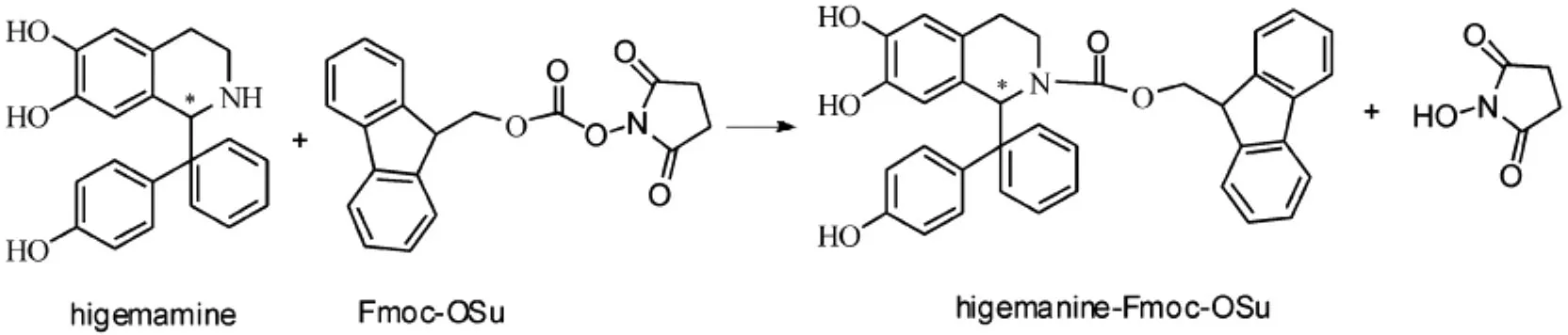
图1 去甲乌药碱与Fmoc-OSu的衍生化反应式
影响去甲乌药碱与Fmoc-OSu衍生化反应的因素有反应介质的酸度、反应时间、衍生化试剂的浓度等.首先,根据文献[7]以硼酸缓冲溶液为反应介质,探讨缓冲溶液的p H值对衍生化效率的影响.从图2可知,硼酸缓冲溶液的p H值为8.5时去甲乌药碱衍生物的峰面积最大,所以本实验将p H值为8.5的硼酸缓冲溶液作为反应介质.其次,在室温条件下,观察反应时间对衍生化反应的影响.图3显示,反应开始时去甲乌药碱衍生物的峰面积随反应时间的增加而逐渐增加,当反应时间达到50 min时,衍生物的峰面积最大且趋于恒定,因此衍生化反应时间选为50 min.最后,考察衍生化试剂浓度对衍生化反应的影响.实验表明,对于浓度为0.04μmol/m L的去甲乌药碱,Fmoc-OSu的浓度为5μmol/m L(衍生化试剂浓度/去甲乌药碱浓度=125)时,衍生物的峰面积最大且趋于恒定,所以将衍生化试剂的比选定为125(Fmoc-OSu浓度/去甲乌药碱浓度).
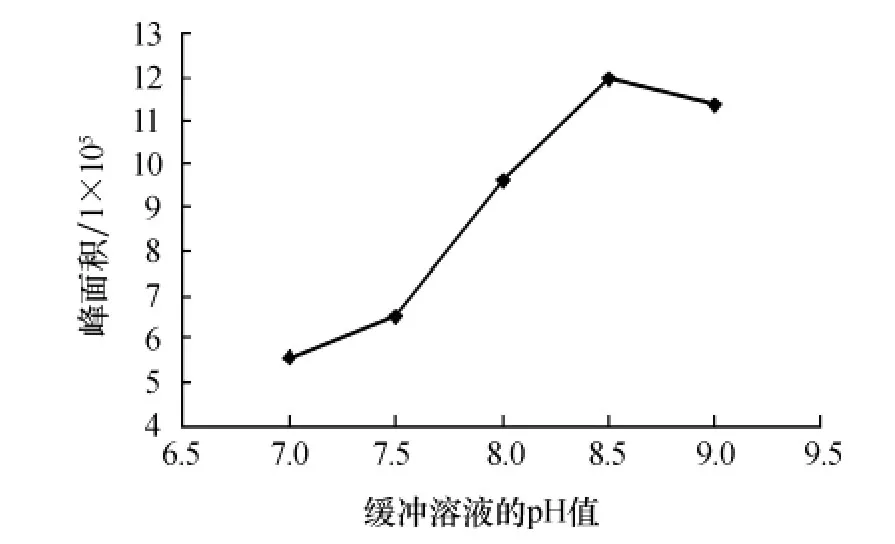
图2 硼酸缓冲溶液p H值对衍生化反应的影响
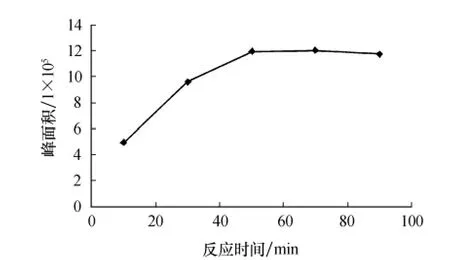
图3 反应时间对衍生化反应的影响
2.2 检测波长及色谱条件
以λ=265 nm为激发波长,λ=315 nm为发射波长,对去甲乌药碱衍生物进行检测.采用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色谱柱,进行衍生化反应液中去甲乌药碱衍生物的分离.在甲醇-水、乙腈-水、磷酸盐缓冲液(p H=3.0)-乙腈、磷酸二氢钠(p H=3.0)-乙腈等洗脱液中,当以乙腈-水(体积分数比为85∶15)作为流动相,流速为1 m L/min时,去甲乌药碱衍生物与基质中的其他峰得到了很好的分离(见图4).根据与空白和样品色谱图的比较,确定保留时间为13.6 min的峰为去甲乌药碱衍生物,其他峰为过量Fmoc-OSu和反应副产物.
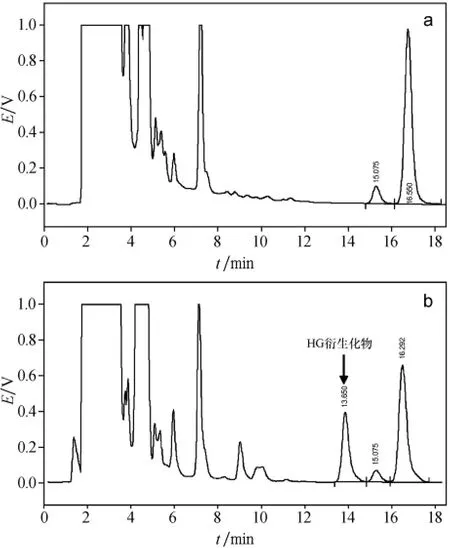
图4 去甲乌药碱与过量Fmoc-OSu衍生化反应后的色谱图:a为Fmoc-OSu(空白),b为HG-Fmoc-OSu衍生物
2.3 线性关系和检出限
去甲乌药碱在0.05~20μg/m L范围内呈良好的线性关系 (r=0.999 8),其回归线性方程为Y=1.181 76×106X+18 320.427 25(Y为去甲乌药碱衍生物峰面积,X 为去甲乌药碱浓度).当S/N=3时,检出限为5.0 ng/m L,比HPLCUV[3]方法高10倍.
2.4 精密度和稳定性
将浓度为10μg/m L的去甲乌药碱标准溶液衍生化后,同1天连续进样5次,置于冰箱(4℃)中的衍生化产物连续5 d进样.结果显示,日内精密度和日间精密度分别为2.74%和9.14%.
2.5 加标回收率
采用标准加入法对分析方法进行准确度验证.取1.25 g莲子心粉末6份,分别加入1.2 mg去甲乌药碱标样,考察样品的加标回收率.结果显示,样品的回收率为88.3%~112.5%,相对标准偏差为5.8%~8.4%(n=5).
2.6 莲子心中去甲乌药碱含量的测定
利用本方法测定莲子心中的去甲乌药碱含量.莲子心提取液的衍生化色谱图见图5.在色谱图中,实验保留时间为13.69 min的峰是去甲乌药碱衍生物,其含量测定结果为94.0 mg/100 g.
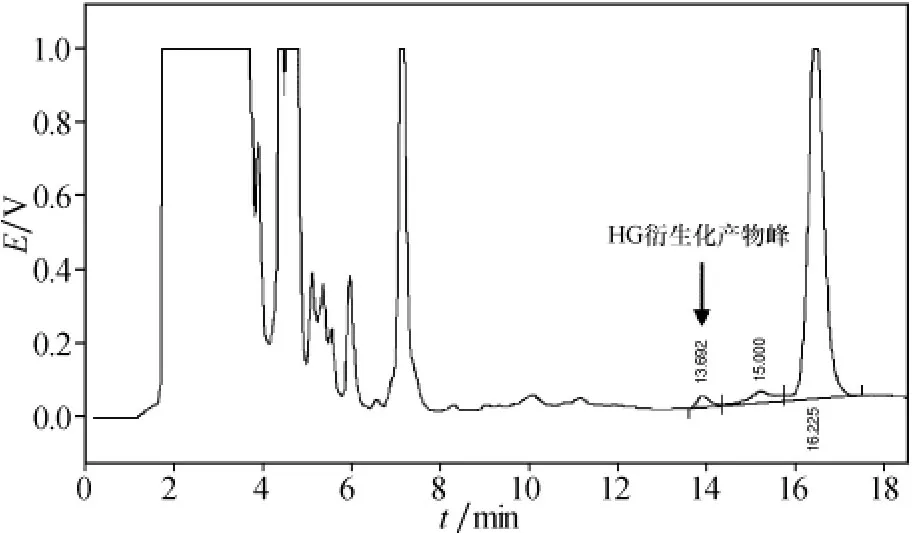
图5 莲子心提取液的衍生化色谱图
3 结论
研究了HPLC-FLD分离去甲乌药碱的Fmoc-OSu柱前衍生化条件和色谱条件.使用该方法可使衍生化反应定量地进行,其反应条件温和,衍生化产物较稳定,而且过量的衍生化试剂和副产物均不干扰待测物的分离和检测.该方法选择性好,检测灵敏度高,可用于含去甲乌药碱的中药材和相关制剂的质量控制.
[1] Kosuge T,Yokota M.Studies on cardiac principle of aconite root[J].Chem Pharm Bull(Tokyo),1976,24:176-178.
[2] 周素娟,杜贵友.去甲乌药碱对心血管系统的影响[J].中国中药杂志,2003,28(10):910-913.
[3] 陈宝玲,郑英丽,陆直,等.HPLC法测定去甲乌药碱的含量[J].中国新药杂志,2004,13(7):628-630.
[4] Lo C F,Chen C M.Determination of higenamine in plasma and urine by high performance liquid chromatography with electrochemical detection[J].J Chromatogr B,1994,655:33-39.
[5] Feng S,Hu P,Jiang J.Determination of higenamine in human plasma and urine using liquid chromatography coupled to positive electrospray ionization tandem mass pectrometry[J].J Chromatogr B,2011,879:763-768.
[6] 丁雅韵,谢孟峡.Fmoc-OSu作为液相色谱分离分析氨基酸的柱前衍生试剂研究[J].分析实验室,2001,20(Suppl):138-139.
[7] Hong H,Lee Y I,Jin D.Determination of R-(+)-higenamine enantiomer in Nelumbo nucifera by highperformance liquid chromatography with a fluorescent chiral tagging reagent[J].Microchemcal J,2010,96:374-379.
Determination of higenamine in Nelumbo nucifera by HPLC with precolumn derivatization
TIAN Hai-feng1, YI Tao2, JIN Dong-ri1*
(1.Department of Chemistry,College of Science,Yanbian University,Yanji 133002,China;2.JILin Tobacco Industrial Co.,LTD,Yanji 133001,China)
A sensitive method for determination of higenamine based on a derivatization reaction with a fluorescent tagging reagent,9-fluorenylmethoxycarbonylsuccinimide(Fmoc-OSu)is developed.The Fmoc-OSu preferably reacts with higenamine under mild reaction conditions in the presence of borate buffer(p H=8.5)to produce the corresponding fluorescent derivative with an excitation maximum at 265 nm and an emission maximum at 315 nm.The derivative of higenamine are efficiently resolved on a UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)column by an isocratic elution with water-acetonitrile(85∶15)mobile phase.The linear range is 0.05-20μg/m L(r=0.999 8)for higenamine.The average recovery of the higenamine is 94.1%-105.9%(RSD 2.74%).The limits of detection(S/N=3)per injection is 5.0 ng/m L.The developed method is applied successfully to the determination of higenamine in embryo of Nelumbo nucifera,a Chinese herbal medicine.
higenamine;precolumn derivatization method;HPLC;Fmoc-OSu;Nelumbo nucifera
O657.72
A
1004-4353(2012)02-0150-04
2012-01-20
*通信作者:金东日(1965—),男,教授,研究方向为药物分析.
