卟啉的生物合成途径与化学合成方法的比较*
2012-09-25苏优拉张逸李嘉宾陆军农
苏优拉 张逸 李嘉宾 陆军农
(1中国药科大学2010届基地班本科生;2中国药科大学2010届制药工程专业本科生;3中国药科大学无机化学教研室 药学基础化学实验中心 江苏南京 211198)
在自然界的生命体中,有一些化合物发挥着非常重要的作用,比如:叶绿素,其介导的光合作用将光能转化为化学能储存于植物体中,是地球上有机体生存和发展的源泉;细胞色素C,能促进氢与氧的结合,加强体内的氧化供能反应,是细胞呼吸过程中电子传递体的主要组成部分;血红素,作为血红蛋白和肌红蛋白的核心结构域,负责氧气和二氧化碳的转运,在生物体的新陈代谢中起着举足轻重的作用。令人惊奇的是,这些化合物虽然在生物体中所处部位不同、所起作用迥异,但是,它们都含有一个共同的核心结构——卟啉。
卟啉是在卟吩环上拥有取代基的一类大环化合物的总称。卟吩是由4个吡咯环和4个次甲基桥联起来的大π共轭体系,其结构如图1所示。天然卟啉类化合物一般是卟吩的吡咯环上的氢被不同基团取代所形成的,例如图1中的血红素、叶绿素和细胞色素C。卟啉的化学合成方法虽然早在1935年就被首次报道,近年来也进行了一系列的改进,但仍存在产率低、产物分离困难、能合成的卟啉种类有限等缺点。本文介绍卟啉的生物合成途径以及近年来的一系列文献报道的化学合成方法,并尝试通过比较分析,找寻它们之间存在的联系。

图1 重要的卟啉类化合物
1 卟啉的生物合成途径
卟啉的生物合成几乎存在于所有真核细胞中,可分为6步(图2),即:①δ-氨基-γ-酮戊酸(ALA)的形成;② 吡咯单元(PBG)的形成;③ 尿卟啉原Ⅲ(含HMB中间体)的形成;④ 尿卟啉原Ⅲ的氧化;⑤ 粪卟啉原Ⅲ的氧化;⑥ 原卟啉原Ⅸ的氧化。其中,第②步缩合、第③步环合和第⑥步氧化涉及卟啉环骨架的构建,而第④步和第⑤步只是对卟啉环上侧链的修饰。
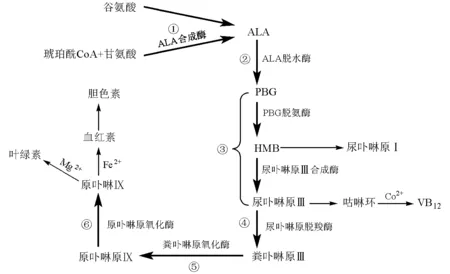
图2 卟啉类化合物的生物合成途径[1-2]
1.1 ALA的形成
ALA的形成是卟啉生物合成中的第一步,也是限速步骤[2]。在生物体中,ALA可由两条途径形成(图3)。Shemin途径[4]由David Shemin于1945年首次发现并逐步完善,主要存在于不进行光合作用的真核生物中,如动物和真菌[5]。ALA合成酶末端含一个赖氨酸(Lys)残基,当没有底物时,辅基磷酸吡哆醛(pyridoxal-5′-phosphate,PLP)与其形成Schiff碱,存在底物甘氨酸(Gly)时,Gly与PLP形成Schiff碱再和琥珀酰CoA缩合形成ALA的同时释放CO2[3]。Beale等于20世纪70年代中期发现了以谷氨酸(Glu)为起始原料的C5途径[6]。它存在于植物、大多数细菌和所有古细菌中。C5途径主要依赖3种酶,连接酶通过形成谷氨酰基-1-tRNA激活1位羧基,还原酶将羧基还原为醛基,再经转氨酶的作用形成ALA[3]。在少数几种生物中也发现两条途径都存在[5]。
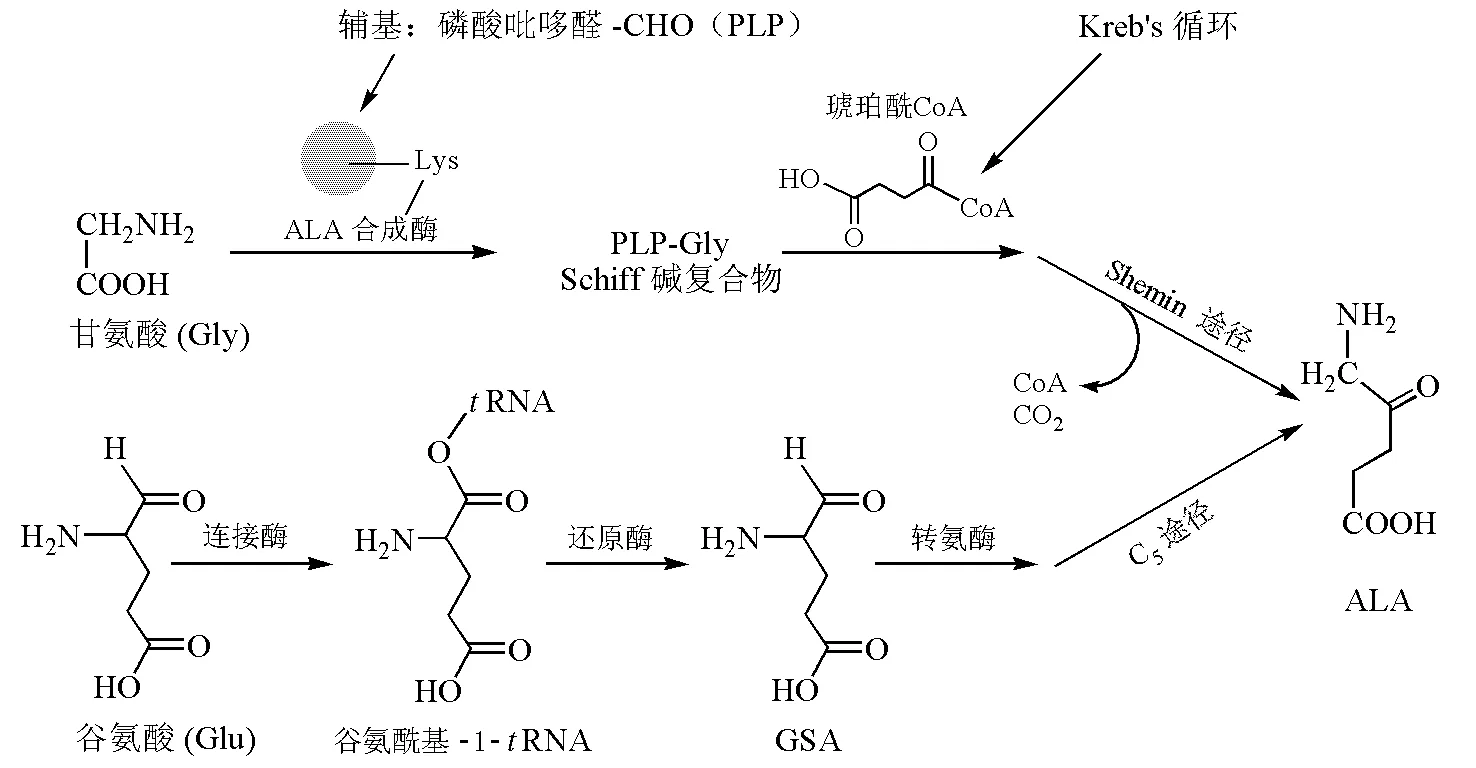
图3 ALA形成的两种途径[3]
1.2 PBG的形成
两分子ALA之间不对称缩合产生第一个吡咯衍生物——PBG(porphobilinogen)[5](图4)。反应机制与Knorr吡咯缩合反应相似,首先,2个ALA分子与酶活性部位的保守Lys残基形成Schiff碱,P位ALA分子的C4和A位ALA分子的C3进行Aldol缩合形成C—C键,接着P位ALA分子的氨基进攻羰基碳原子形成C—N键[7]。
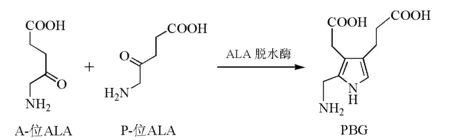
图4 两分子ALA 缩合形成PBG
1.3 HMB的形成及其转化为尿卟啉原Ⅲ
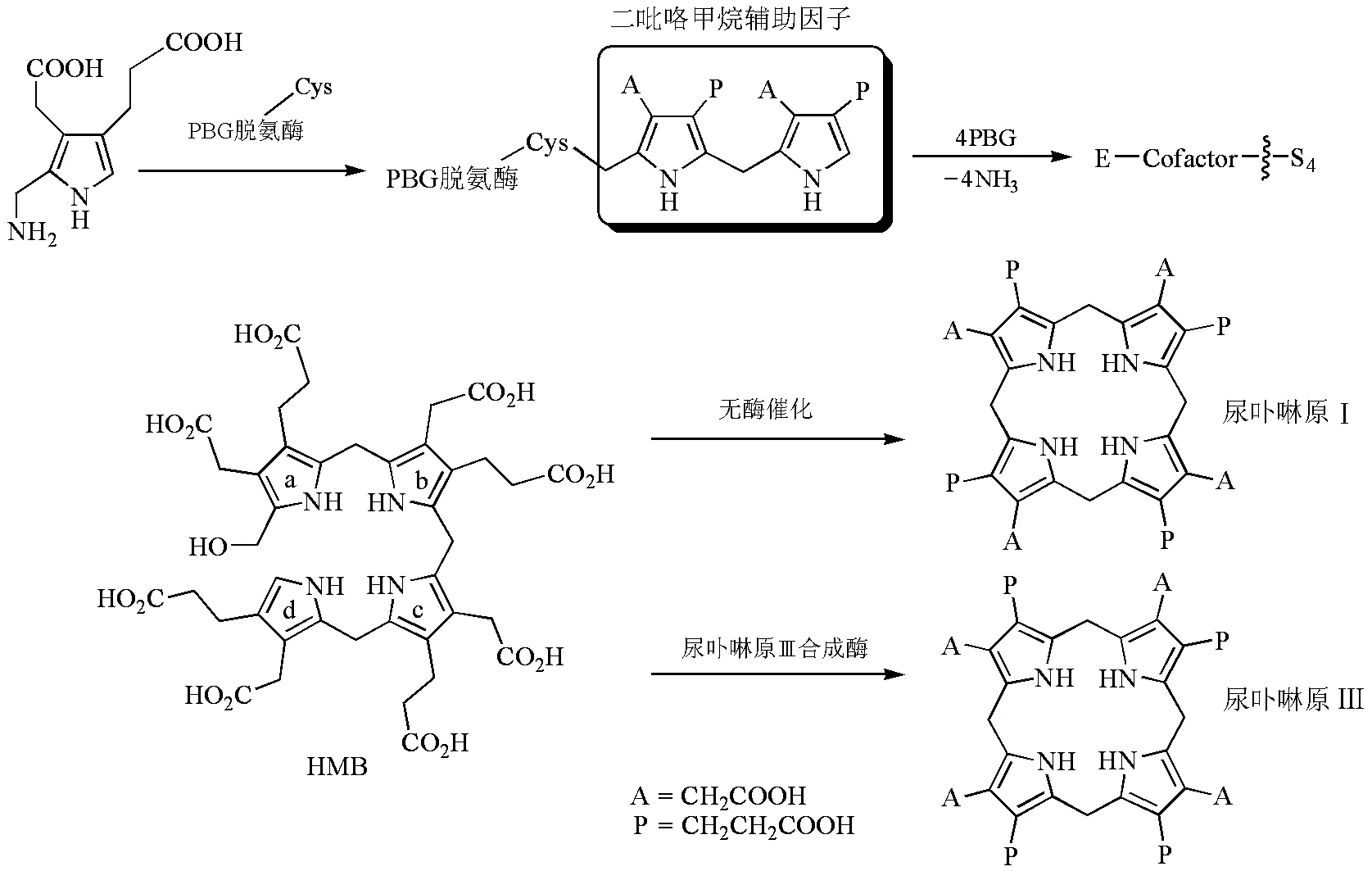
图5 尿卟啉原的形成(HMB中间体)[3,10,13]
4分子PBG缩合形成尿卟啉原Ⅲ(uroporphyrinogen Ⅲ)是卟啉环生成的一个关键步骤,很多人对其具体过程提出了不同的猜想[8-10]。1980年Battersby A.R.等通过同位素标记证明了HMB(1-hydroxymethylbilane,羟甲基胆色烷)是尿卟啉原Ⅲ合成过程中的中间体;1987年他们又发现了新型辅基——二吡咯甲烷(dipyrromethane,DPM)[11],为阐明具体步骤奠定了基础。尿卟啉原Ⅲ的形成由两种酶共同完成(图5)。首先,脱氨酶将4个PBG组装形成开链HMB;在此过程中,先合成出二吡咯甲烷辅助因子,与脱氨酶末端的半胱氨酸巯基以硫醚键相结合,DPM再和4分子底物相连接;当DPM上连有4个吡咯单体时,a环和DPM之间的键断裂形成HMB。第二步,HMB被转运到尿卟啉原Ⅲ合成酶上,在环合的同时,d环重排生成尿卟啉原Ⅲ[12]。在无尿卟啉原Ⅲ合成酶时,HMB在酸催化下迅速形成有毒性的不被代谢的尿卟啉原Ⅰ。值得注意的是,编码两种酶的基因位于同一操纵子内,两个基因协同表达[5],但尿卟啉原Ⅲ合成酶的表达量远远超过脱氨酶,以保证在生理条件下总是生成尿卟啉原Ⅲ[8]。
1.4 尿卟啉原Ⅲ—粪卟啉原Ⅲ—原卟啉原Ⅸ
尿卟啉原脱羧酶(uroporphyrinogen Ⅲ decarboxylase,UROD)和粪卟啉原氧化酶(coproporphyrinogen Ⅲ oxidase,CPOs)催化卟啉环侧链的修饰(图6)。在生理底物浓度下,4个环的脱羧作用是按d→a→b→c的顺序发生;当底物浓度超过生理浓度时,脱羧作用以随机方式发生[5]。脱羧后生成的粪卟啉原Ⅲ(coproporphyrinogen Ⅲ)在哺乳动物中,经过位于线粒体外膜的依赖O2的CPOs催化,最终把电子传递给O2。此过程不需要金属和辅助因子辅助[2],CPOs的催化机理至今仍不清楚[5],详细过程见文献[14]。
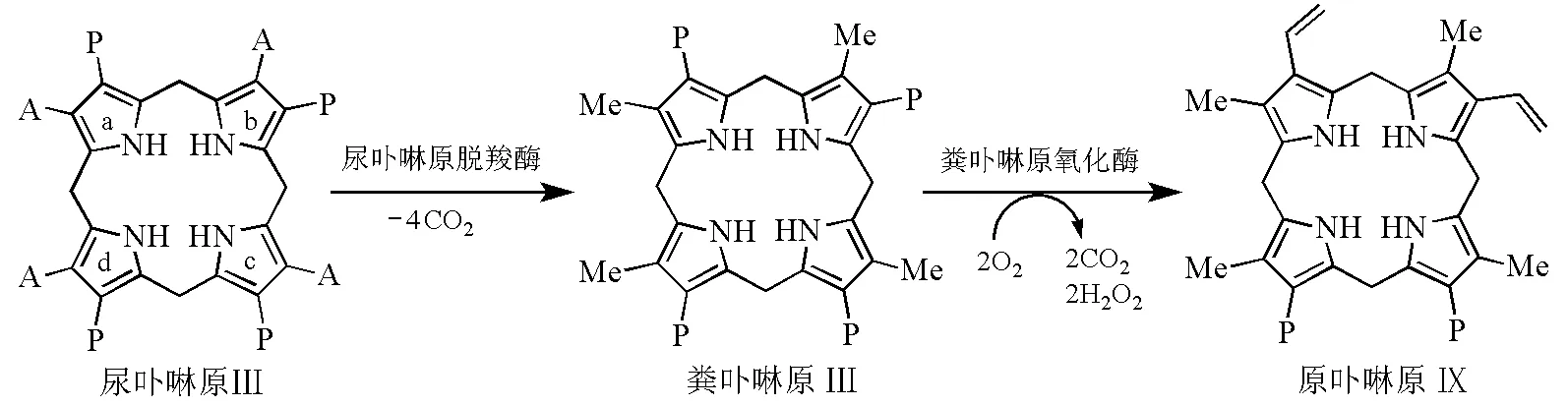
图6 卟啉环侧链的修饰[2]
1.5 原卟啉原Ⅸ的氧化[15]
原卟啉原氧化酶(protoporphyrinogen Ⅸ oxidase, PPOs)位于线粒体内膜的外表面,以O2作为最终的电子受体,以FAD为辅助因子,催化原卟啉原Ⅸ(protoporphyrinogen Ⅸ)的氧化,最终形成完全共轭的大环体系(图7)。生成的原卟啉Ⅸ(protoporphyrin Ⅸ)被直接转运到亚铁螯合酶上,以避免其对细胞的损害(原卟啉Ⅸ对光高度敏感,在O2存在下,经光照射会产生自由基)。
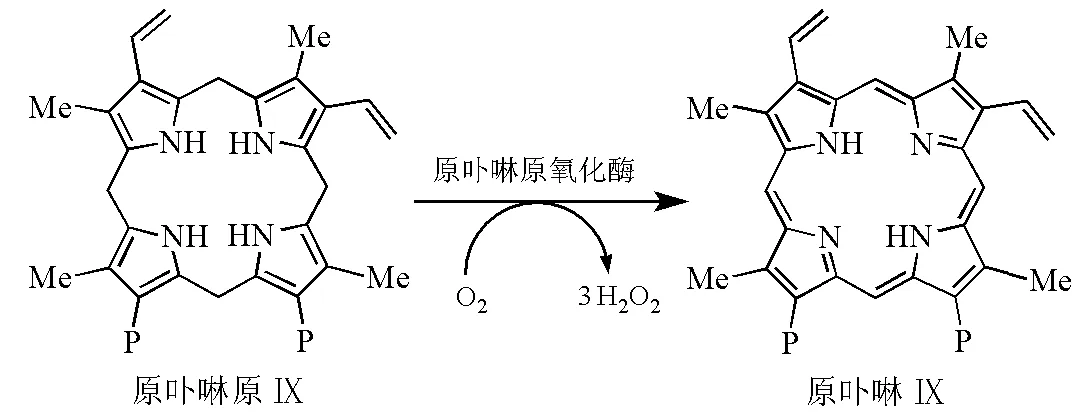
图7 原卟啉原Ⅸ的氧化
2 卟啉的化学合成方法总结
目前,卟啉的化学合成方法主要有两种:① 4个吡咯单体直接缩合环化生成卟啉(简称四吡咯合成法);② 模块法。合成方法和路线的选择取决于目标卟啉分子的结构特点,中位对称取代的卟啉主要用四吡咯合成法,而不对称卟啉、天然卟啉及其类似物主要采用模块法合成。
2.1 四吡咯合成法
2.1.1 Rothemund法
卟啉类化合物最早由Rothemund合成[16]。Rothemund法以醛类化合物(甲醛、乙醛、苯甲醛等)和吡咯为原料,以吡啶和甲醇为溶剂在封管中反应,90~95℃下反应24~48h(图8)。该法反应时间长,所需反应条件苛刻,而且后处理非常麻烦,产率很低;在此条件下,能用来作反应物的取代苯甲醛极少[17]。
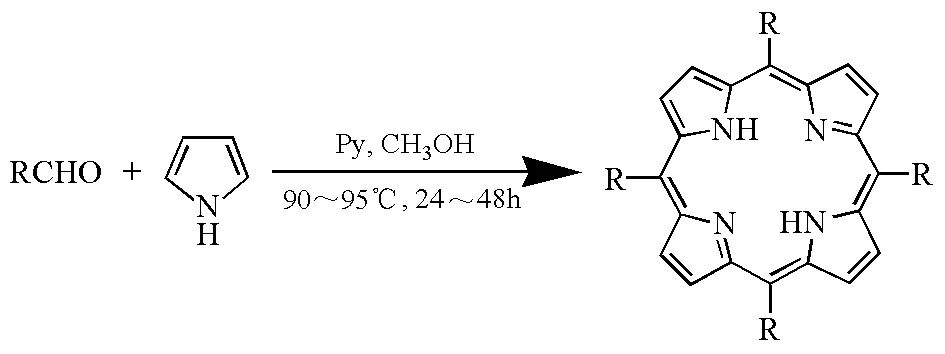
图8 Rothemund法
2.1.2 Adler-Longo法及其改进
Adler和Longo等以有机质子酸作催化剂成功地制备了卟啉,并在1964年提出了卟啉生成的反应机理[18]。该法采用苯甲醛和吡咯在丙酸中回流反应30min,经冷却、过滤、洗涤及真空干燥,得到四苯基卟啉,产率达20%(图9)。此法的优点是操作比较简单,实验条件不算苛刻,产率较高。但由于反应条件的限制,一些带敏感基团的取代苯甲醛不能用作原料,带有强吸电子基的苯甲醛为底物时产率特别低;反应极易产生大量焦油状物,导致纯化非常困难;另外反应中的副产物四苯基二氢卟啉与四苯基卟啉分离较困难[17]。
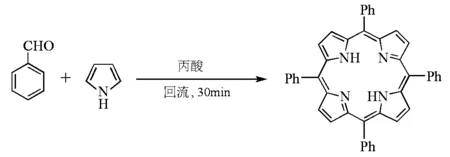
图9 Adler-Longo法
潘继刚等[19]对Adler-Longo法作了进一步调整,他们采用催化量的有机酸和极性溶剂代替丙酸介质,反应过程中产生的杂质明显减少,四苯基卟啉的产率最高达到50%。研究溶剂和催化剂对反应的影响,发现H+在反应过程中起催化剂的作用,pKa2.0~4.0的酸作催化剂,合成产率较高。以二甲苯、甲苯、氯苯、硝基苯、苯甲醚为溶剂,四苯基卟啉产率较高,可达30%~50%。
2.1.3 Lindsey法及其改进
Lindsey等[20]进一步改进了四苯基卟啉的合成,采用苯甲醛和吡咯在氮气保护下,在二氯甲烷中,以三氟化硼和乙醚络合物催化,整个反应分两步进行,先得到卟啉合成的中间体卟啉原(porphyrinogen),然后,以二氯二腈基苯醌(DDQ)或四氯苯醌(TCQ)将卟啉原氧化得到最终产物卟啉,从而使反应可以在常温下进行。近20年来,Lindsey小组进一步研究了此一锅两步合成法的影响因素,发现酸催化剂的种类和用量、醛和吡咯上的取代基以及反应物浓度均会影响反应产率,并对主要副产物——链状聚合物的组成进行了分析[22-24]。由于Lindsey法的反应温度较低,较少产生焦油状副产物,目标产物的分离提纯较容易;同时较低的反应温度也允许反应物先经过化学修饰,连接上一些敏感基团,平均产率可达45%~50%(图10)。但该反应浓度低,且最大反应容积为1L,放大后效果不好[21]。反应条件较苛刻,需要无水无氧操作,且反应还不能一步生成四苯基卟啉,必须在反应过程中另外加入氧化剂。
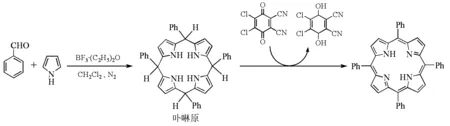
图10 Lindsey法
郭灿城等[25]采用N,N-二甲基甲酰胺为溶剂,无水AlCl3为催化剂,苯甲醛和吡咯缩合生成四苯基卟啉,产率可达30%,高于Adler法。反应过程中不需氮气保护,产物不含副产物四苯基二氢卟啉,并且反应时间也较短,为2h。该方法的应用范围较广,对于以取代苯甲醛为原料的合成反应,产率在25%~30%之间。缺点是催化剂AlCl3易与水反应,给产物的分离造成困难。
Adler-Longo法和Lindsey法是目前应用比较广泛的两种方法。它们的主要区别在于所用的催化剂不同,Adler-Longo法用有机质子酸作为催化剂,而Lindsey法则用Lewis酸作为催化剂。
2.1.4 微波催化合成法
1986年,Gedye等[26]发现微波可显著加快有机合成反应速率。从此,微波在合成化学领域迅速得到重视。以二甲苯为溶剂,对硝基苯甲酸为催化剂使苯甲醛和吡咯在微波炉内反应20min,可以得到卟啉,产率为9.5%[27]。微波辅助合成卟啉在我国发展较快,研究表明微波作用的时间与强度、溶剂及催化剂的选择、反应试剂的组成及用量等均对卟啉的合成有较大的影响。此法避免了传统加热合成方法的反应时间较长(一般需回流2~3h)、副反应多、产率不高且产物难提纯等缺点,且符合节能环保、绿色化学的发展趋势[28]。
2.2 模块法
模块法主要包括[2+2]和[3+1]两种方法。
[2+2]合成亦称MacDonald方法[29], 即两分子二吡咯甲烷缩合产生卟啉母核。该法可方便地合成具有C2对称轴的四苯基卟啉,也称为trans-卟啉,还可合成中位是4个不同芳基取代的卟啉。近年来,随着原料二吡咯甲烷衍生物合成方法的逐步改进和优化,可以合成的衍生物种类逐渐增多,产率也得到了提高,使[2+2]法的应用范围越来越广[30]。
[3+1]法是由MacDonald方法衍生出来的,将一个由两个桥碳原子连接的3个吡咯环组成的胆色素分子和一分子a,a′-二甲酰基吡咯环合得到卟啉的合成方法。此方法总产率较低,能合成一些结构复杂且较为特殊的卟啉,主要用于扩充卟啉的种类[31]。
关于卟啉化学合成方法更详细的总结可参考文献[32]。
3 化学合成和生物合成之间的联系
3.1 起始原料
化学合成和生物合成两种途径都以单个吡咯环作为合成卟啉环的起始原料。不同的是,化学合成通常以吡咯或取代吡咯以及醛类为原料,而生物合成途径中由两个ALA分子通过类似Knorr吡咯缩合产生的带亚甲氨基侧链的吡咯单元(PBG)作为起始原料。
3.2 单体吡咯的聚合方式
在生物合成和化学合成过程中,吡咯都是通过质子化、脱氨或脱水、再去质子的3步循环逐步加成形成聚合物。在此过程中都会产生活泼烯键,以促进吡咯的聚合(图11)。在生物合成中,吡咯单元(PBG)自身带有亚甲氨基侧链(来自甘氨酸),侧链脱氨之后形成烯键而被活化;而在化学合成中则是吡咯进攻醛基,产生带羟基的侧链,羟基质子化脱水后形成烯键而被活化。
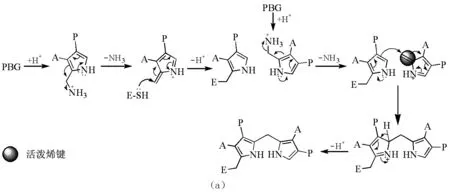

图11 单体吡咯聚合的可能机理(a)生物合成;(b)化学合成
3.3 链状吡咯聚合物环合形成卟啉原
两种途径都是单体吡咯先聚合成链状吡咯聚合物,然后4个吡咯单体经亚甲基桥再进一步环合成卟啉原(图12)。在生物合成途径中,PBG脱氨酶与辅助因子DPM和吡咯聚合物形成复合物,当连有4个吡咯时,环a和酶之间的键会水解断裂形成链状HMB。其原因至今仍是一个谜,可能与酶的空间结构和新型辅助因子二吡咯甲烷有关;可能类似于生物体内多糖和多肽的形成过程。
根据Adler等提出的机理,化学合成时吡咯和醛类先形成长短不一的链状聚合物,推测反应中形成的焦油状副产物很可能是吡咯和醛类的链状聚合物或者吡咯自身的聚合物。主要生成环状四聚体可能与卟啉独特的空间结构的稳定性有关。
在环合时,生物合成途径有一步特有的环翻转过程,HMB的亚甲基被活化后,进攻与其距离较远的C16,与C16环合形成螺中间体,d环翻转,碳链在另一处断裂再环合形成尿卟啉原Ⅲ。而化学合成没有环的翻转过程,活泼烯键直接与C19环合形成卟啉环,与体内尿卟啉原Ⅰ的形成过程类似。
3.4 卟啉原氧化形成大环共轭的卟啉
在生物合成中,原卟啉原Ⅸ在PPOs的催化下形成大环共轭的卟啉。PPOs以二聚体形式存在,由3个结构域组成(图13),分别为FAD结合域、底物结合域和膜结合域。原卟啉原氧化酶的辅助因子FAD发挥重要的电子传递作用,以O2作为最终的电子受体,因其每次只能转移两个电子,所以要经过3个独立的循环才能氧化完全,产生二氢卟啉和四氢卟啉中间体(图14)。当酶与底物结合时,底物带负电荷的丙酸基侧链与酶的精氨酸残基结合,使底物以一定方向被固定在酶上,只能以a环和d 环间的亚甲基桥通过酶的狭缝与FAD的N5原子接触,环上的氢原子通过亚胺-烯胺互变异构重排并逐步被氧化。
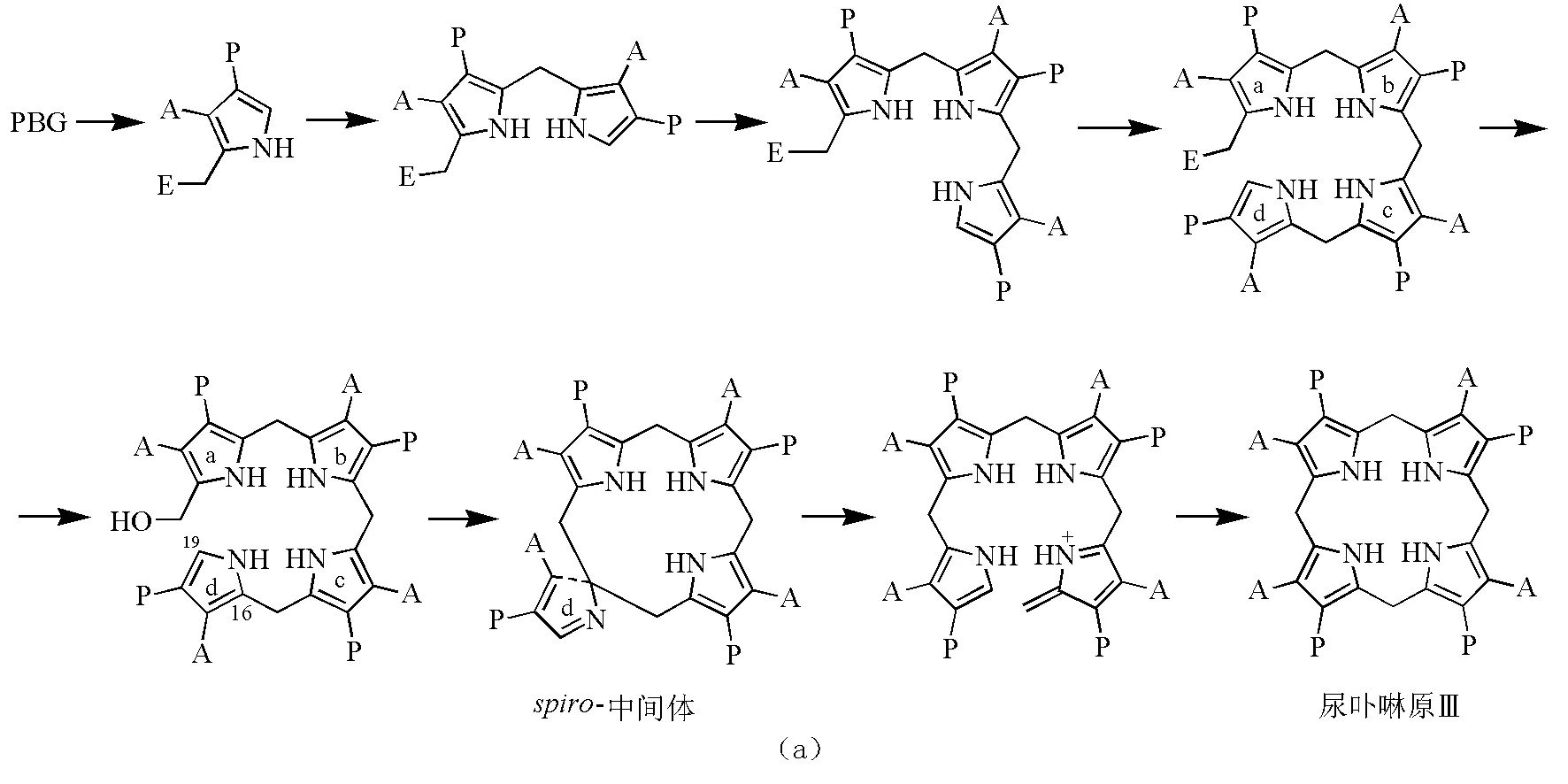
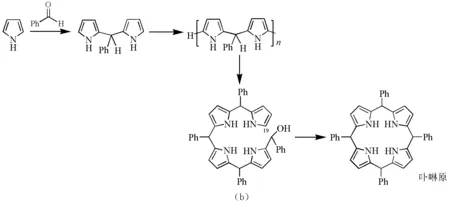
图12 由吡咯单体形成卟啉原的过程(a)生物合成;(b)化学合成
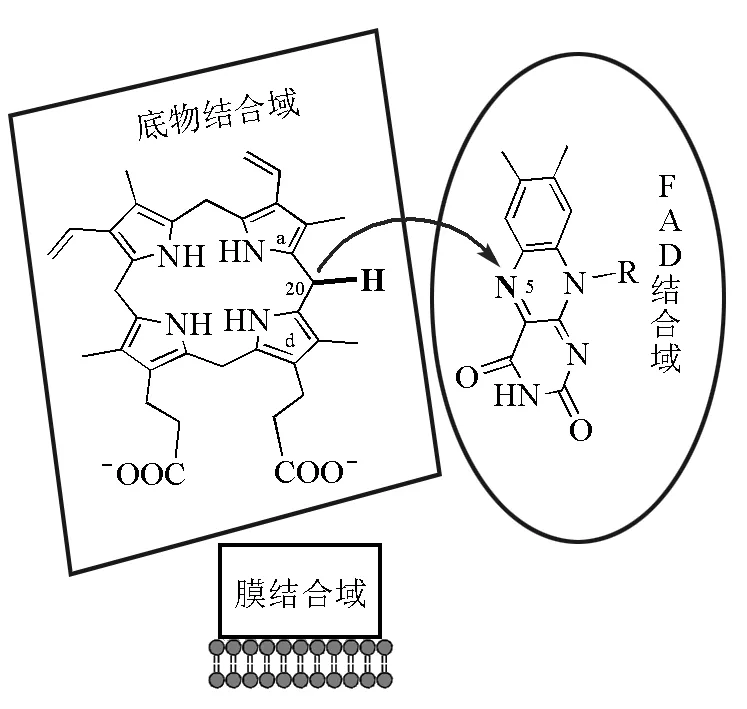
图13 原卟啉原氧化酶的空间结构[15]
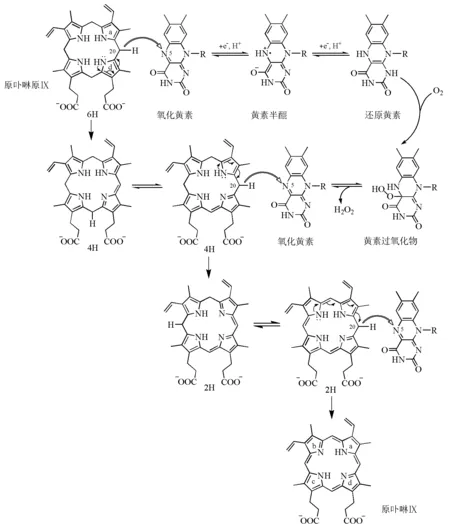
图14 生物合成中原卟啉原Ⅸ的氧化[18,33-34,41]
在化学合成中,4分子醛与吡咯缩合,最终环合后产生卟啉原。1970年,Dolphin等证实卟啉原确实是卟啉合成过程的中间体[31]。卟啉原不稳定,极易被氧化,但在一般条件下氧化又不完全,产生的二氢卟吩混在卟啉中很难除去[35]。在反应中加入氧化剂或在反应结束后再进一步氧化能提高产率。硝基苯[36]、DMSO[37]、DDQ[20]以及最近文献报道的MnO2[38]和SeO2[39]等均能作为氧化剂,实现从卟啉原到卟啉的氧化,但氧化反应的具体机理至今仍不太清楚。
在卟啉原的氧化过程中,生物合成和化学合成都是通过互变异构转移H—N和H—C,逐步延长共轭链,产生二氢卟啉和四氢卟啉的中间体并最终形成大环共轭的卟啉。不同的是在生物合成过程中,底物被酶包裹,由于酶对底物的固定作用,卟啉原上所有的氢必须都转移到C20上才能被氧化;而且由于酶蛋白对底物的稳定作用,中间体二氢卟啉和四氢卟啉可以以不太稳定的形式存在。而在化学合成过程中,氧化反应可以在任一亚甲基桥上发生,而且氧化后氢转移的最终结果要保证共轭链的延伸,中间体二氢卟啉和四氢卟啉要以相对稳定的形式存在。
4 总结和展望
近年来,由于卟啉类化合物独特的分子结构,在仿生学、药物化学、分析化学、光物理与化学、材料化学、电化学、催化化学等领域中被广泛地研究与应用,但是较低的化学合成产率限制了进一步发展。本文通过比较卟啉的生物合成途径和化学合成方法,尝试寻找二者的联系,希望能为改进化学合成方法、提高合成产率提供一些启示。
生物合成和化学合成一样,也遵循基本的化学反应规律,反应的本质是相同的。只不过由于酶的参与,酶的辅基或酶上的某个保守基团与底物相互作用,固定或增强底物的反应活性,稳定中间体的结构,降低反应的活化能,可使化学反应高效专一地进行。在化学合成中,影响产率的主要是底物的活性、对聚合度的控制以及卟啉原的氧化;而生物合成在这3方面的控制很精密。基于上述生物合成与化学合成的联系与差别,可以尝试从以下几方面对化学合成进行改进。
(1) 在生物合成过程中,底物的反应活性较高。例如,吡咯聚合时,氨基被质子化后经脱氨酶作用迅速形成活泼烯键,易于被进攻。而在化学合成中,活泼烯键的形成要通过脱水,而在一般条件下自动脱水比较困难。在底物上引入其他适当的离去基团可以促进缩合,有时甚至可以省去最后一步的氧化,直接生成卟啉。Pierre Martin小组2010年报道的[2+2]法就是在底物上引入碘原子,缩合时消除碘化氢和水,不用加氧化剂直接在室温下生成大环共轭的卟啉[40]。此方法的原理若能推广至其他卟啉合成方法中,有可能显著降低反应温度,提高产率。
(2) 生物合成途径对副产物的控制非常严格,原料利用率较高。吡咯聚合时一端通过巯基与酶相连,只有另一端延伸。当尿卟啉原Ⅲ合成酶催化链状吡咯聚合物的环合时,将聚合度严格控制在4,不会生成长链吡咯聚合物或者多元环状吡咯聚合物。这一点若化学合成则实现起来较困难。而且,目前的化学合成都需要酸催化,吡咯在酸性条件下容易因聚合而被破坏。抑制吡咯自身的聚合并精确控制吡咯和醛类的链状聚合物的聚合度尚有待于进一步改善。可以先在酸催化下不断延伸生成长链聚合物,将聚合物的一端固定,再用PBG脱氨酶和尿卟啉原Ⅲ合成酶从另一端进行切割和组装;或者在反应中加入具有空腔的材料,而空腔的大小恰好能容纳四聚体,这样就能把反应分割成多个单元,抑制聚合物的形成。例如,各种型号的分子筛孔径大小不同,具有一定的酸性,又能吸水,可以尝试。此外,既然卟啉能和金属离子形成配合物,可以模拟冠醚的合成方法,通过金属离子的模板效应来促进四聚体的形成。
(3) 生物合成途径的最后一步为芳构化反应,在辅助因子FAD的作用下,以氧气作为最终的电子受体,实现了大π共轭体系(即卟啉环)的构建。在化学合成模拟生物合成时,聚合和氧化这两步分别进行,以避免在聚合过程中生成氧化的中间体,阻碍四聚体的形成。最近有文献报道分别以MnO2和SeO2作为氧化剂,用于卟啉原的氧化,能提高产率。它们虽然易于后处理,但都会污染环境。继续寻找价格低廉且环境友好的氧化剂,温和并高效地完成芳构化反应,符合当前绿色化学的发展趋势和要求。
[1] Scott A I.JOrgChem,2003,68(7):2529
[2] Ajioka R S,Phillips J D,Kushner J P.BiochimicaetBiophysicaActa,2006,1763:723
[3] Porra R J.PhotochemPhotobiol,1997,65(3):492
[4] Battersby A R.NatProdRep,2000,17:507
[5] Heinemann I U,Jahn M,Jahn D.ArchBiochemBiophys,2008,474:238
[6] Beale S I.ProcNatlAcadSciUSA,1975,72(7):2719
[7] Leeper F J.NatProdRep,1985,2(1):19
[8] Frydman B,Frydman R B.AccChemRes,1975,8(6):201
[9] Scott A I,Ho K S,Kajiwara M.JAmChemSoc,1976,98(6):1589
[10] Battersby A R,Mcdonald E.AccChemRes,1979,12(1):14
[11] Hart G J,Miller A D,Battersby A R,etal.JChemSoc,ChemCommun,1987,23:1762
[12] Battersby A R.JNatProd,1988,51(4):629
[13] Leeper F J.NatProdRep,1985,2:561
[14] Jackson A H,Elder G H,Smith S G.IntJBiochem,1978,9(12):877
[15] Koch M,Breithaupt C,Messerschmidt A,etal.EMBOJ,2004,23:1720
[16] Paul R.JAmChemSoc,1935,57:2010
[17] 郝晓伶,韩士田,刘彦钦.河北师范大学学报(自然科学版),2009,33(1):85
[18] Adler A D,Longo F R,Williams H.JAmChemSoc,1964,84(15):3145
[19] 潘继刚,何明威,刘轻轻.有机化学,1993,13(5):533
[20] Lindsey J S,Schreiman I C,Hsu H C,etal.JOrgChem,1987,52(5):827
[21] 杨彪.精细化工,1999,16:56
[22] Geier Ⅲ G R,Lindsey J S.JPorphyrinsPhthalocyanines,2002,6:159
[23] Geier Ⅲ G R,Lindsey J S.JChemSoc,PerkinTrans2,2001(5):677
[24] Geier Ⅲ G R,Lindsey J S.Tetrahedron,2004,60:11435
[25] 郭灿城,何兴涛,邹纲要.有机化学,1991,11(4):416
[26] Gedye R.TetrahedronLett,1986,27(3):279
[27] Petit A.SynthCommun,1992,22(8):1137
[28] 汉玉霞,韩士田,刘彦钦.化学工程与装备,2008,6:98
[29] Arsenault G P,Bullock E,Macdonald S F.JAmChemSoc,1960,82:4384
[30] Lindsey J S.AccChemRes,2010,43(2):300
[31] 王周锋,邓文礼.化学进展,2007,19(4):520
[32] akthitharan S,Edwards C,Boyle R W.Tetrahedron,2000,56:1025
[33] Massey V.BiochemSocTrans,2000,28(4):283
[34] Mattevi A.TrendsBiochemSci,2006,31:276
[35] 王君文,何明威.化学试剂,2001,23(1):9
[36] 杨琴,冯清.中国药物化学杂志,2006,16(3):154
[37] 章艳,高保娇.合成化学,2008,16(1):86
[38] Bruno F O N,António M R G,Marta P.InorgChemCommun,2010,13:395
[39] Stephanie M S L,Diogo R B D,Eugênia R D,etal.TetrahedronLett,2011,52:1441
[40] Pierre M,Markus M,Dietmar F,etal.OrgProcessResDev,2010,14:799
[41] Banerjee R.REDOX BIOCHEMISTRY.Hoboken,New Jersey:Wiley John & Sons,Inc,2008
