气相色谱法测定樟酚酊中樟脑含量
2012-09-14陈刚
陈 刚
(湖南省张家界市食品药品检验所,湖南 张家界 427000)
樟酚酊系张家界市人民医院制剂注册品种,协定处方,由樟脑、苯酚、甘油、乙醇等组方,具有消炎、止痒等功效,主要用于预防和治疗皮肤瘙痒症。该品种原质量标准比较简单[1],仅规定了性状、化学鉴别等检验项目,未制定含量测定检测指标。为较好地控制其内在质量,笔者采用气相色谱(GC)法对樟酚酊中樟脑进行含量测定,结果准确、重现性好,现报道如下。
1 仪器与试药
GC-2010型气相色谱仪(日本岛津),AOC-5000型自动进样器,GCsolution色谱工作站;TM-WAX(天美)毛细管柱(30 mm ×0.32mm,0.25μm);AUW220D 型电子分析天平(日本岛津),1020a超纯水器(重庆摩尔),KQ2200DB型数控超声波清洗器(昆山超声仪器公司)。樟脑对照品(批号为 110747-201008,纯度 97.0%,供含量测定用),苯酚对照品(批号为100509-200401,供鉴别用),水杨酸甲酯(批号为 110707-201011,内标物,纯度99.9%),均来自中国药品生物制品检定所;高纯氮气(批号为20110329,纯度99.999%,长沙日臻气体有限公司),水为超纯水,其他试剂为分析纯;樟酚酊样品3批(批号分别为20110901,20110929,20110930,张家界市人民医院制剂室)。
2 方法与结果
2.1 色谱条件
以 TM-Wax毛细管柱(30mm×0.32mm×0.25μm)为分析柱,FID检测器,分流进样,分流比40∶1,进样口温度:230℃,柱温:125℃,检测器温度:250℃。樟脑峰和内标物质峰的分离度应大于 1.5。
2.2 内标溶液制备
精密称取水杨酸甲酯1.139 39 g(纯度99.9%),精密称定,置25mL容量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀,作为内标溶液。
2.3 对照品溶液制备和校正因子测定
精密称取樟脑对照品0.021 19 g(纯度97.0%),置25mL容量瓶中,精密加入内标溶液1mL,用无水乙醇稀释至刻度,摇匀。吸取1μL注入气相色谱仪,记录峰面积,计算校正因子。
2.4 供试品溶液制备
精密量取本品2 mL置50 mL容量瓶中,精密加入内标溶液2 mL,用无水乙醇稀释至刻度,摇匀,即得。
2.5 方法学考察
阴性干扰试验:按樟酚酊处方组成除不加樟脑外,制成缺“樟脑”的阴性样品,照供试品溶液同法制成阴性对照品溶液。精密吸取对照品溶液、供试品溶液及阴性对照品溶液各1μL,分别注入气相色谱仪,测定。结果阴性对照品溶液色谱图中,在樟脑峰相应的保留时间附近,无干扰峰检出。气相色谱图见图1。

图1 气相色谱图
线性关系考察:精密称取樟脑对照品 0.234 71 g(纯度97.0%),置25mL容量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀,作为对照品贮备溶液。另精密称取水杨酸甲酯0.991 14 g(纯度99.9%),置25mL容量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀,作为内标贮备溶液。精密量取对照品贮备溶液1,2,3,5,7mL分别置25mL容量瓶中,各瓶精密加入内标贮备溶液1mL,用无水乙醇稀释至刻度,摇匀。分别吸取1μL,注入气相色谱仪。以樟脑与内标物的峰面积之比为纵坐标(Y)、樟脑质量浓度为横坐标(X),得线性回归方程 Y=0.906 39X -0.005 88,r=0.999 9(n=5),线性范围为 0.364 3 ~2.549 9 g/L。
精密度试验:精密吸取对照品溶液1μL,按上述色谱条件进样,重复6次,测定对照品与内标物峰面积的比值。结果的 RSD为0.46%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取同一供试品(批号为20110901)溶液1 μL,按上述色谱条件,分别在 0,2,4,8,12,24 h 时进样,测定樟脑和内标物的峰面积,计算校正因子,以内标法计算含量。结果的 RSD为0.13%(n=6),表明供试品溶液在24 h内基本稳定。
重现性试验:取同一批号(20110901)的樟酚酊样品6瓶,分别精密吸取2mL,照供试品溶液制成6份供试液,按上述色谱条件测定樟脑含量。结果的 RSD=0.23%(n=6),表明方法精密度高,重现性较好。
加样回收试验:精密称取樟脑对照品 0.223 49 g(纯度97.0%),精密称定,置10mL量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀,作为对照品贮备溶液(21.678 53 g/L)。再精密称取水杨酸甲酯 0.990 15 g(纯度99.9%),精密称定,置25 mL量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀,作为内标贮备溶液(39.566 39 g/L)。另取已知含量的样品[批号为 20110901,含量为标示量的 99.63%(19.926 g/L)]0.5,1,2mL 共 6 份,置 50mL量瓶中,分别精密加入对照品贮备溶液(21.678 53 g/L)0.5,1,2mL,再分别精密加入内标贮备溶液1mL,用无水乙醇稀释至刻度,摇匀,分别吸取1μL,注入气相色谱仪,记录峰面积。根据标准曲线计算樟脑的含量和回收率。结果见表1。
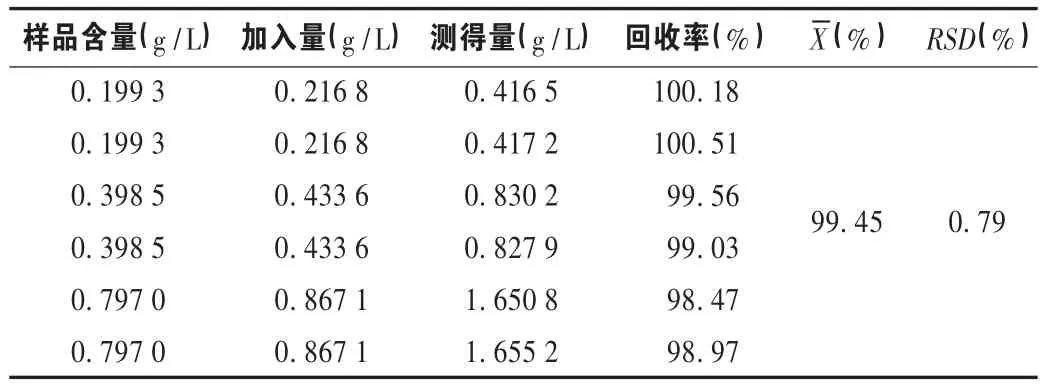
表1 樟脑加样回收试验结果(n=6)
2.6 样品含量测定
取样品 3 批(批号为 20110901,20110929,20110930),按供试品溶液制备方法制成供试液,精密吸取1μL,注入气相色谱仪,测定樟脑和内标物的峰面积,以内标法计算含量。结果3批样品中樟脑含量分别为标示量(100mL∶2 g)的99.63%,99.48%,99.54%。
3 讨论
曾有报道应用高效液相色谱法测定樟脑含量的方法[2]。由于气相色谱法具有分离速度快、效率高、保留时间短、试验成本相对较小等优点,故对于既可用液相又可用气相时,一般考虑首选气相色谱法。
改变柱温,可改善分离效能。文中所述色谱条件未采用程序升温,樟脑与溶剂(乙醇)、内标物等成分能达到较好分离。经试验,按上述色谱条件采用程序升温(柱温为120℃保持5min,以10℃ /min的速度升至160℃,保持2 min),结果樟脑和内标物(水杨酸甲酯)也能达到较好分离,樟脑峰保留时间在3~4min左右,可满足含量测定要求。两种方法均适用的原因是本品药物组分较少,分离容易。若是复杂组分的样品,应以程序升温法较好。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:1 108.
[2]余小平,舒金富,黄 华,等.高效液相色谱法同时测定樟脑苯酚溶液中两组分的含量[J].药物分析杂志,2006,26(8):1 161-1 162.
