浓香型白酒窖泥中细菌多样性的免培养技术分析*
2012-09-12汤斌刘金英周庆武李安军万春环汤有宏
汤斌,刘金英,周庆武,李安军,万春环,汤有宏
1(安徽工程大学微生物发酵安徽省工程技术研究中心,安徽芜湖,241000)
2(安徽古井贡酒股份有限公司,安徽毫州,236826)
浓香型白酒窖泥中细菌多样性的免培养技术分析*
汤斌1,刘金英1,周庆武2,李安军2,万春环2,汤有宏2
1(安徽工程大学微生物发酵安徽省工程技术研究中心,安徽芜湖,241000)
2(安徽古井贡酒股份有限公司,安徽毫州,236826)
采用免培养(culture independent)技术直接从浓香型白酒窖泥中提取细菌微生物混合基因组DNA(也叫元基因组DNA),利用细菌16S rDNA通用引物扩增窖泥细菌的序列,根据16S rDNA序列对细菌多样性进行初步分析。采用PCR扩增技术、分子克隆技术以及序列同源性分析等方法测定细菌的16S rDNA,通过与基因数据库中相似菌群序列同源性的比较,得到样品菌种多样性,分别构建系统发育树。结果显示,细菌含有几大类群,表现出高度的细菌多样性。共分为Uncultured bacterium、Clostridium、Lactobacillus、Eubacterium和 S yntrophomonas五个细菌分类。
浓香型白酒,窖泥,微生物多样性,免培养法,系统发育树
近年来分子生物学技术不断兴起,研究者可以选用更先进的分子技术手段分析样品的各种信息资源。16S rDNA分类方法被大家广泛采用,将免培养分析法与其结合,能准确快速的分析样品中微生物构成的多样性[1-3]。
中国浓香型白酒的生产以泥窖的窖池为基础,发酵的过程就是栖息在窖泥和糟醅的庞大而复杂的微生物相互之间做用的结果。由于我国研究专家在固态发酵窖池上具有得天独厚的地理优势,在窖泥的微生物生态,特别是在主要功能菌的分离培养,人工培育活性窖泥等方面取得了很大的成绩。但仅仅依托于传统的微生物分离培养的分析方法是远远不能满足窖泥微生物生态学研究的需要。众多研究表明自然界微生态系统中可培养的微生物仅仅占微生物总数的0.1%~10%[4]。本研究运用免培法初步探索了白酒窖池窖底窖泥中细菌的多样性构成,通过窖泥细菌菌群16S rDNA序列同源性和系统发育远近关系初步了解了窖泥中含有哪些微生物,初步了解细菌的组成框架,希望对窖泥的传统培养法提供一点指导意义。
1 材料与方法
1.1 实验材料
(1)分析样品:安徽古井贡酒酒厂窖池底层窖泥。
(2)菌种:DH5α Escherichia coli,安徽工程大学可再生资源研究室保藏。
(3)主要试剂和仪器:Taq DNA聚合酶、PCR产物纯化试剂盒,上海生工;连接酶及相关试剂,Takara公司;DNA大片段回收试剂盒(Biotech),抗生素氨苄青霉素,上海生工;PCR引物合成,上海生工;英国Techne公司的TC-312型PCR仪;高速冷冻离心机,贝克曼公司;XSJ-HS型显微摄影仪,上海华宇。
1.2 实验方法
1.2.1 总DNA的提取
从土壤样品中高效提取可进行分子操作的基因组DNA是进行免培养法的基础。此过程包括:从环境样品中高效获得粗制基因组DNA和去除粗制DNA中的杂质。
直接提取窖泥微生物总DNA的方法采用预处理和不进行预处理的提取方法有明显区别:未预处理的提取方法得到的DNA样品电泳条带有明显的弥散现象,而经过预处理得到的DNA样品的条带较整齐。这是因为对窖泥进行预处理可以有效地去除杂质,减少胞外游离DNA污染,减少可溶性的无机物、有机物和腐植酸类物质的污染。特别是腐殖酸影响后续PCR的质量所以在参考大量的文献[5-9]后本研究采用如下方法:取适量的窖泥冷冻干燥过夜;取200 mg加入2 mL EP管中;采用0.1 mol/L pH7.6磷酸盐缓冲溶液(NaCl 0.8%,KCl 0.02%,Na2HPO40.15%,KH2PO40.025%)1 mL浸泡5 min;1000 r/min离心2 min,去上清液;重复步骤2依次;加入0.35 g无菌玻璃珠(直径1 mm)以及0.8 mL DNA提取液(100 μmol/L Tri,100 μmol/L EDTA,200 μmol/L NaCl,3%CTAB,pH 9.0),剧烈振荡10 min加入溶菌酶500 μL(500 mg/mL)混匀37℃水浴30 min(期间颠倒混匀3次);(4)加入0.8 mL SDS缓冲液(100 mmol/L Tris,200 mmol/L NaCl;3%SDS,pH9.0)振荡混匀;65℃水浴30 min(期间颠倒混匀3~5次),10000 r/min离心10 min收集中间液相层(分层后将上层液相全部取出);加入等体积氯仿/异戊醇(24∶1)抽提2次12000 r/min离心5 min取上清液加入1/10体积的醋酸钠溶液混匀,加入等体积的异丙醇混匀;室温沉淀30 min以上;最大转速离心15 min取沉淀,70%乙醇洗涤室温自然晾干,溶于适量ddH2O中。用大片段DNA回收试剂盒纯化去除杂质,-20℃保存。
1.2.2 总DNA质量的检测
用紫外分光光度计检测DNA在A260、A280波长下的值,核酸的紫外最大吸收值在260 nm,蛋白质的紫外最大吸收峰在280 nm,利用核酸和蛋白质的这个特性,一般用A260/A280表示DNA样品纯度。一般A260/A280≈1.8较纯,否则可能存在蛋白质或小分子杂质等污染[10]。
将总DNA样品用1.2%(w/v)琼脂糖凝胶在电压100 V,电泳1 h条件下电泳,EB染色,于紫外凝胶成像拍照系统观察。
1.2.3 窖泥PCR扩增
用细菌16S rDNA通用引物[11]F:5’-AGAGTTTGATCCTGGCTCAG-3'和 R:5'-AAGGAGGTGATCCAGCCGCA-3'进行扩增。PCR扩增反应体系总50 μL,模板2 μL。PCR反应条件为:95℃预变性5 min,然后94℃变性1 min,55℃退火50 s,72℃延伸50 s,循环30次,72℃延伸10 min。
1.2.4 PCR产物纯化
去除dNTP、引物二聚体和非特异性核酸片段等杂质用DNA胶回收纯化试剂盒进行切胶纯化。
1.2.5 PCR产物的克隆以及鉴定
用胶回收试剂盒对扩增出的目的片段进行纯化,产物连接到pUCT载体,转化到DH5α中,运用蓝白斑原理筛选阳性克隆,将转化产物涂布到筛选的LB平板(含有氨苄、IPTG和X-gal),待蓝白斑长出,用牙签挑取白色菌落于含氨苄的LB液体培养基中。待培养基变浑浊,初步鉴定为克隆成功。再利用菌液PCR法进一步鉴定是否是阳性克隆,用电泳检测,将含有DNA片段的菌液送交测序公司。
1.2.6 基因的序列测序和分析
阳性克隆送到上海生工测序,去除序列头尾中所含的载体序列,根据图谱去除尾部信号杂带,余下序列进行拼接。提交序列到NCBI基因库中进行序列同源性比对,找到各序列的同源性较高的16Sr DNA序列,根据同源相似性的比值对序列进行分类。同时对这些序列用ClustalX软件[12]按照最大同源性的原则进行多序列比对,根据比对结果用DNAsp、MEGA3.1、PAUP等软件转化格式用邻接法((Neighbor-joining method)构建系统发育树[13],以确定其系统发育地位。进一步分析窖泥细菌多样性。图3是基于PAUP软件构建的NJ型系统发育树。
2 结果与分析
2.1 DNA样品提取结果分析
图1的1~6号泳道为窖泥总DNA用琼脂糖凝胶电泳后的条带,M为2000bp的分子量标记。可见6个样品的DNA目的片段杂质少且条带清晰。由于基因组模板的纯度会影响后续实验结果[14]。同时将样品用紫外分光光度仪定性检测DNA的纯度。选取A260/A280最接近1.8的样品进行后续的PCR扩增。
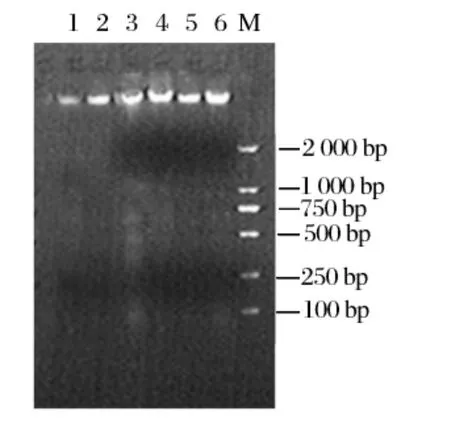
图1 窖泥DNA样品琼脂糖凝胶电泳结果
2.216S rDNA基因的PCR扩增结果
图2长度约1.5 kb的目的片段条带清晰。图2是以窖泥细菌总DNA为摸板,利用细菌通用引物扩增出的16S rDNA基因片段,是切胶纯化后的结果。如图条带清晰,可以进行后续的克隆实验。
2.3 窖泥16S rDNA序列同源性与系统发育树分析
本研究中提取窖泥样品6份DNA样品,分别进行PCR反应,每次PCR设置3次重复,将3次重复的PCR产物混合后利用PCR产物回收试剂盒回收,进行后续实验。然后将6份DNA样品的PCR产物充分混合后分别连接在载体上,用于克隆。利用这种方法可以大大增加减少PCR过程中的可能存在的误差。本研究共得到30个有效的阳性克隆,将有效的16S rDNA基因的全部序列上传到网络数据库NCBI做同源性比对和序列分析,同源性上获得的总序列中有11个序列为未培养菌株,19个序列为鉴定出的细菌序列。其中有3个序列与已报道的未培养或未鉴定菌株的16S rDNA序列的同源相似性≥97%,8个序列与已报道的未培养或未鉴定菌株的16S rDNA序列的同源相似性在94%~96%;另有5个序列与已知的序列相似性为97%~100%,14个序列与已知细菌的序列相似性在91%~96%。一般全序列具有99%~100%相似性的判定为一个种,全序列在97%~99%相似性的认定为一个属[15-16]。对于同源性上有22个克隆子与网络数据库NCBI做同源性比对低于97%。可见浓香型白酒窖泥中存在许多未知的菌种等待开发。
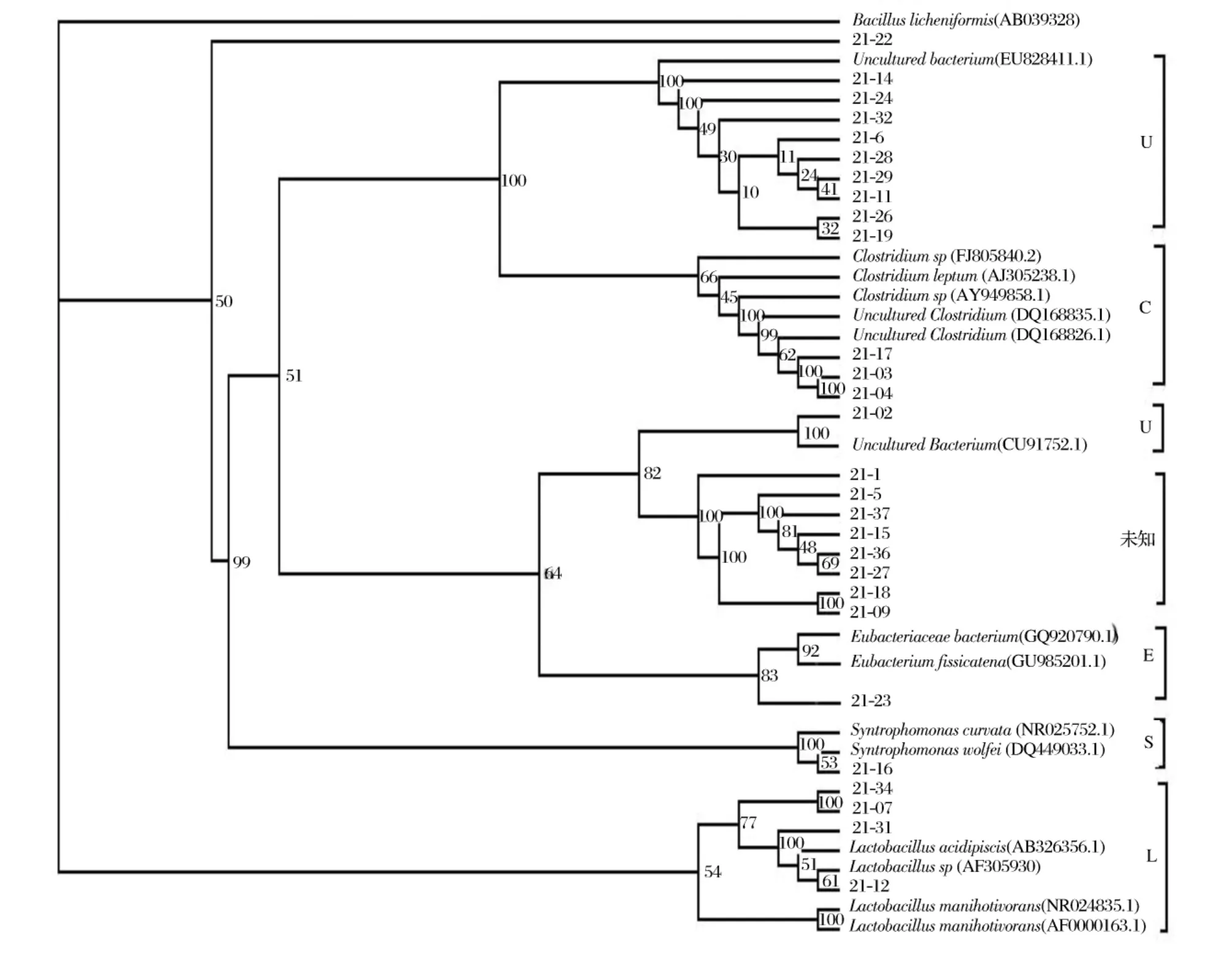
图3 基于16S rDNA序列为基础的窖泥中细菌的系统发育树图(NJ树)
从系统发育树结构看,以Bacillus licheniformis为外群构建,结构分为8个部分。有效克隆分别属于Uncultured bacterium、Clostridium、Lactobacillus、Eubacterium和Syntrophomonas 5个细菌分类。克隆子21-22和图3结构中未知部分的8个克隆分化成独立的结构,表明与已知菌种的几个属的进化距离较远,他们无疑代表了新的分类单元,如果能深入研究这些序列的宿主菌对深入探索窖泥细菌多样性更具有深远的意义。
对于整个细菌域来说,除了未培养菌株外,梭状芽孢菌为窖泥的优势菌,并没有呈现多种类的多样性。这可能由于窖泥底层,土壤结构、营养成分、理化性质、土壤环境很稳定,一些适应此环境的微生物就形成较优势的菌群生存,不适应这个环境的微生物就逐渐被淘汰。Clostridium类群能降解有机底物产生醇类、有机酸、CO2/H2以及矿物质成分,一般从厌氧的窖泥中分离,在白酒窖泥中与甲烷菌共生,并在中后期游离于糟醅中,难于单独分离培养或不能培养,包括一些产己酸菌、丁酸菌等[17],可与淀粉质物质水解产生己酸、丁酸并形成己酸乙酯、丁酸乙酯等有关。浓香型白酒主要呈香成分是己酸乙酯,所以梭状芽孢杆菌起到了很重要的作用。
Lactobacillus类乳酸类细菌菌群为第二优势菌,适合于在厌氧环境下的繁育,白酒发酵中乳酸属的主要代谢产物为乳酸,是形成乳酸乙酯的主要物质,乳酸菌同时会产生乙酸、乙醇、CO2等其它代谢产物,丙酸、丁酸等菌可以利用乳酸为底物或为碳源形成成丙酸或丁酸,形成白酒其它香味物质成分。在白酒发酵初期乳酸菌起到调节酸度的作用,发酵中期为其它微生物提供必需氨基酸和维生素促进酿酒微生物的繁殖,发酵后期乳酸乙酯的不挥发特性,留在酒尾中增加了酒风味中的后味。整个发酵过程中乳酸菌的发酵产物多,代谢方向多,又作为多种微生物的底物,表明其在白酒发酵中至关重要的地位。
Syntrophomonas类互营单胞菌和Eubacterium类在系统发育树中独立成一分枝。在整个克隆子中也分别仅有一个克隆子属于这两类。他们在白酒研究中很少报道,需要研究者进一步研究其在白酒生产中的相关作用。
本研究得到的细菌菌群多样性结构,与已有窖泥文献报到存在共同点同时也存在一些差异。传统的微生物培养法和新兴的分子生物技术都检出窖泥中有梭状菌、乳酸菌[18-21],本次应用免培养法检出都含有这些菌。表明这两种菌群对白酒发酵起到至关重要的作用。差异性则是Syntrophomonas类互营单胞菌和Eubacterium类优杆菌很少有相关报道,另外,对于免培养法发现的很多序列只和未培养和未鉴定的菌种序列相似,需要对这些序列更进一步的研究才能把握其在白酒发酵中的作用。对研究结果的相似性和差异性,一方面浓香型白酒呈香成份主要为己酸乙酯所以主要的菌种类群基本相似,另一方面由于各酿酒厂土质、地理环境的差异以及窖泥时间长短等因素的不同;再者免培养研究法本身所具有的不可避免的方法缺点所以肯定存在差异性。
3 讨论
本文利用免培养法对安徽古井贡酒酒厂窖池窖底的窖泥进行细菌微生物多样性的研究,来确定窖泥中优势细菌的组成情况。结果共有Uncultured bacterium、Clostridium、Lactobacillus、Eubacterium和Syntrophomonas 5个细菌分类。但免培养法存在以下几个缺陷:1)由于免培养法是以PCR反应为基础的,所以由于混合样品中的不同DNA与载体亲和度的不同对结果会产生影响,使得亲和度大的片度较易反映,而某些基因片度却不能鉴定出。这是方法本身存在的缺陷,在实验时采用把多次提取的基因组样品进行混合后进行多组PCR,以减少误差。2)对混合样品进行克隆,存在重复克隆的现象。本文挑取的克隆数不多,但初步反应了细菌的多样性组成,在排除假阳性后获得有效基因序列仍然是细菌组成的一个缩影。所以将获得的有效克隆全部送去测序,通过序列结果可以看出克隆重复度。以后为避免方法的缺陷,需将克隆数加大,更可能全面的反应细菌构成。该方法的好处在于免于对微生物的长时间培养,快速省时尤其是生化反应不明显、难以培养的细菌;采用分子水平的鉴定也有比较高的准确度和可信度,尤其适用于及传统表型方法难以鉴定出的细菌。
窖池窖底的窖泥的细菌结构组成可以通过本方法大致了解。因为窖池底层结构基本没有太大变化,对菌种的要求比较苛刻,利于形成优势菌群。了解窖池底层的菌群结构更有代表性的了解某一地理环境下的窖池窖泥。随着分子技术水平的不断发展,要确定窖泥中微生物的精确构成,确定新物种的产生,对新物种代谢和产物的研究,仅靠本文的研究方法是肯定不够的,这就要结合传统的技术和新型的研究方法,以排除单一方法存在的缺陷,做出更细致准确的研究成果。
[1]Amann R I,Ludwig W,Schleifer K H.Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J].Microbiological Reviews,1995,59(1):143-149.
[2]Giovannoni S J,Britschgi T B,Moyer C L,et al.Genetic diversity in Sargasso Sea bacterioplankton[J].Nature,1990,345(6270):60-63.
[3]Ward D M,Weller R,Bateson M M,et al.16S rRNA sequences reveal numerous uncultured microorganisms in a natural community[J].Nature,1990,345(6270):63-65.
[4]Brock T D.The study of microorganisms in situ:progress and problems[A].Symposia of the Society for General Microbiology[C],Midsion,1987,41:1-17.
[5]Zhou J Z,Bruns M A,Tiedje J M.DNA Recovery from soils of diverse composition[J].Applied and Environmental Microbiology,1996,62(2):316-322.
[6]赵勇,周志华,李武,等.土壤微生物分子生态学研究中总DNA的提取[J].农业环境科学学报,2005,24(5):854-860.
[7]曹治明,郑维,权春善.土壤DNA提取方法的研究[J].大连民族学院学报,2005,7(3):94.
[8]陈敏.土壤样品中DNA提取方法的比较[J].微生物学杂志,2005,25(3):101-104.
[9]宋培勇.从土壤中提取DNA方法比较[J].微生物学杂志,2006,26(1):109-112.
[10]Versalovic J,Koeuth T.Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes[J].Nucleic Acids Research,1991,19(24):6823-6831.
[11]Edwards U,Rogall T,Blocker H,et al.Isolation and direct complete nucleotide determination of entire genes characterization of a gene coding for 16S ribosomal RNA[J].Nucleic Acids Research,1989,17(19):7843-7853.
[12]Thompson J D,Gibson T J,Plewniak F,et al.The ClustalX windows interface:Flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25:4876-4882.
[13]Tamura K,Dudley J,Nei M,et al.MEGA4:Molecular Evolutionary Genetics Analysis(MEGA)software version 4.0[J].Molecular Biology and Evolution,2007,24:1596-1599.
[14]Frostegard A,Courtois S,Ramisse V,et al.Quantification of bisa related to the extraction of DNA directly from soils[J].Applied and Environmental Microbiology,1999,65:5409-5420.
[15]Drancourt M,Bollet C,Carlioz R,et al.16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates[J].Journal of Clinical Microbiology,2000,38(10):3623-3630.
[16]Janda J M,Abbott S L.Bacterial identification for publication:when is enough enough?[J].Journal of Clinical Microbiology,2002,40:1887-1891.
[17]姚万春,唐玉明,任道群,等.优良窖泥功能菌的筛选及其生物学特性的初步研究[J].酿酒科技,2010(11):33-35.
[18]乔宗伟,张文学,张丽莺,等.浓香型白酒发酵过程中酒醅的微生物区系分析[J].酿酒,2005,32(1):32-34.
[19]邓依,唐云容,张文学.16-23S RNA ITS-AFLP指纹图谱分析在白酒窖泥细菌多样性分析中的应用[J].酿酒科技,2010(3):46-48.
[20]张文学,乔宗伟,向文良,等.中国浓香型白酒窖池微生态研究进展[J].酿酒,2004,31(2):31-35.
[21]唐云容,钟方达,张文学,等.浓香习酒窖泥微生物菌群多样性及系统发育分析[J].酿酒科技,2011(12):24-28.
ABSTRACTCulture independent method was used to study the diversity of Pit Mud bacteria.The techniques included culture independent approach,PCR technique and 16S rDNA sequence homology analysis etc.The total DNA directly extracted from Pit Mud was used as PCR template.Random clones containing almost full size 16S rDNA sequences(about 1.5 kb)were sequenced and subjected to an on line similarity search.According to the sequences alignment and homology analysis,the bacteria of Pit Mud were consisted of Uncultured bacterium,Clostridium,Lactobacillus,Eubacterium,and Syntrophomonas.
Key wordsGujing-flavor Liquor,Pit Mud,microbiology diversity,culture independent approach,phylogenetic analysis
Phylogenetic Diversity Analysis of Bacteria In Gujing-flavor Liquor Pit Mud Using Culture Independent Method
Tang Bin1,Liu Jin-ying1,Zhou Qing-qu2,Li An-jun2,Wan Chun-huan2,Tang You-hong2
1(Anhui Polytechnic University,Engineering Technology Research Center of Anhui Microbial Fermentation,Wuhu 241000,China)
2(Anhui Gujing Distillery Company Limited,Haozhou 236826,China)
学士,教授。
安徽省自然科学基金(No.KJ2007A018)。
2012-02-28,改回日期:2012-04-27
