HPLC法测定蒙药材蒙酸模(霍日根-其和)中大黄酚的含量
2012-09-12张润祥王志君
张润祥 王志君 丁 宁
(1.呼和浩特市食品药品检验所,内蒙古 呼和浩特 010020;2.内蒙古食品药品检验所,内蒙古 呼和浩特 010070)
蒙酸模原植物载于18世纪蒙医伊希巴拉珠尔著《认药白晶鉴》。19世纪蒙药学家占布拉道尔吉著《无误蒙药鉴》载:“根、茎红色叶大圆形,粗糙,铺地而生,根和茎色红外其他与大黄相同,但根具多皱,种子三角形。”功能:杀“粘”,下泻,消肿,愈伤。用于“粘”疫,痧疾,丹毒,乳腺炎,腮腺炎,骨折,金伤[1]。因此,为了考察该药材的质量,本研究建立了其有效成分大黄酚的HPLC含量测定方法。
1 仪器与试剂
日本岛津2010AHT高效液相色谱仪、日本岛津LC-20A高效液相色谱仪:紫外及二极管阵列检测器,岛津LC-Solution色谱工作站。甲醇为色谱纯;其他均为分析纯试剂;水为纯化水;大黄酚对照品(购于中国药品生物制品检定所,批号:110796-200716)。蒙酸模(收集于呼和浩特市黄花村、平顶村、呼消喜来,共计3批供试品)。
2 实验方法与结果
2.1 色谱条件:色谱柱:AlltimaTM -C18柱(250×4.6mm,5μm);流动相:甲醇—0.1%的磷酸溶液,梯度洗脱[0 ~10min,甲醇 65%;10 ~ 25min,甲醇 65% →80%;25 ~45min,甲醇 80%];检测波长:430nm;柱温:35℃;流速:1.0 mL·min-1。
2.2 对照品溶液的制备:精密称取大黄酚对照品适量,加甲醇制成每1mL含16μg的溶液,即得。
2.3 供试品溶液的制备:取本品粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇25mL,称定重量,加热回流1h,放冷,再称定重量,用甲醇补足减失重量,滤过。精密量取续滤液5mL,减压回收溶剂至干,残渣加8%盐酸溶液10mL,超声处理2min,再加三氯甲烷10mL,加热回流1小时,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷洗涤3次,每次10mL,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇使溶解并转移至10mL量瓶中,加甲醇至刻度,摇匀,即得。
2.4 专属性考察:精密吸取甲醇空白溶剂,大黄酚对照品溶液,供试品溶液各10μL注入液相色谱仪。大黄酚对照品色谱峰峰纯度好,紫外光谱最大吸收为430nm;供试品溶液中色谱峰保留时间与大黄酚对照品一致,在430nm处有最大吸收;甲醇空白溶剂无吸收。表明该含量测定方法专属性好,空白溶剂无干扰。(色谱图见图1、2、3)
图1 空白溶剂色谱图
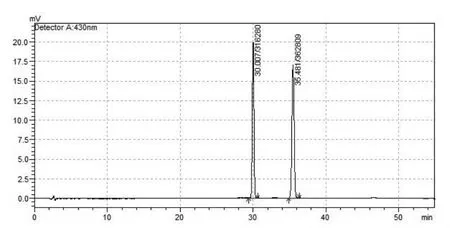
图2 大黄酚对照品色谱图(2号峰)
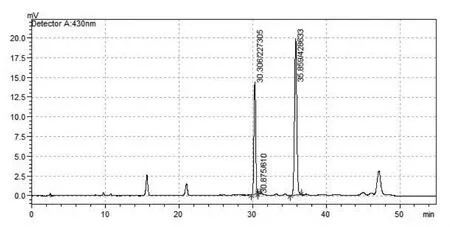
图3 供试品色谱图
2.5 线性关系考察:按“2.2”对照品溶液制备方法称取大黄酚对照品3.173mg,置200mL量瓶中,加甲醇溶解并稀释至刻度(浓度为15.86μg·mL-1)。分别精密吸取对照品溶液 1.0、2.0、5.0、10.0、15.0、20.0、25.0、30.0mL,注入高效液相色谱仪,测定峰面积;以峰面积为纵坐标(Y),进样量为横坐标(X)做线性回归,回归方程为Y=2748.91358 X - 748.56387,r=0.9999。结果表明,大黄酚在15.865 ~475.950μg范围内呈良好的线性关系。
2.6 稳定性试验:取同一份供试品溶液,分别在0,2,4,6,8,10,12h进样测定,RSD 为1.51%。
2.7 重复性试验:取同一批样品按供试品溶液制备方法制备6份样品溶液,分别注入液相色谱仪测定峰面积,RSD为0.96%。
2.8 精密度试验:吸取同一批供试品溶液,在上述色谱条件下重复进样6次,RSD为1.22%。
2.9 加样回收率试验:取同一批已知含量的样品6份,每份取约0.25g(正常量的一半),精密称定,分别精密加入大黄酚对照品适量,按供试品溶液制备方法制备样品溶液,同法操作,分别注入液相色谱仪测定含量,计算回收率,结果见表1。
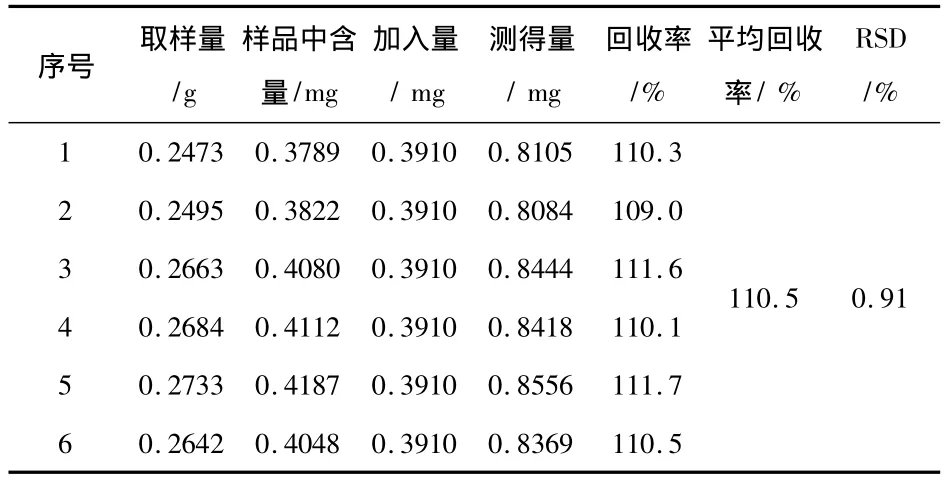
表1 回收率试验
2.10 样品测定:分别取供试品各2份,以上述方法进行分析,大黄酚含量(按干燥品计算)分别为:0.238%、0.135%、0.157%。
3 讨论
检测波长的选择采用岛津LC-20A二极管阵列检测器,在200~600nm波长范围内进行光谱扫描,结果大黄酚在254、430nm波长处均有最大吸收,但430nm波长处杂质峰干扰较少,故选择430nm波长处作为检测波长。
[1]卫生部药典委员会.卫生部药品标准.蒙药分册[S].1998.46.
[2]国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2010.22、1118、1171.
