肝豆状核变性11例临床分析
2012-08-02陈荟冰
陈荟冰,蒋 黎
(1.重庆市公共医疗救治中心六病区,重庆 400030;2.西南医院传染科,重庆 400038)
肝豆状核变性又称Wilson病,是一种常染色体隐性遗传的铜代谢障碍性疾病,导致致病基因为ATP7B,定位于染色体13q14.3,编码一种铜转运P型ATP酶。ATP7B基因突变导致ATP酶功能减弱或丧失,使血清铜蓝蛋白合成减少以及胆道排铜障碍,蓄积于体内的铜离子在肝、脑、肾、角膜等处沉积,引起进行性加重的肝硬化、椎体外系症状、精神症状、肾损害及角膜色素(K-F环)等。肝豆状核变性好发于青少年,男比女稍多。为了提高对Wilson病临床特征的认识,现将2008年1月至2011年9月西南医院感染科住院确诊为肝豆状核变性的11例患者的临床症状、体征及相关理化检查资料分析如下:
1 临床资料
2008年1月至2011年9月西南医院感染科住院的11例肝豆状核变性患者,男性7例,女性4例。年龄最大15岁,最小6岁,中位年龄10岁。诊断标准[1]:(1)锥体外系症状、体征或/及肝病症状体征。(2)血清铜蓝蛋白小于200 mg/L。(3)肉眼或裂隙灯证实有K-F 环。(4)家族遗传史。(5)肝铜>250 μg/g(干重),为诊断该病的金标准,对不能明确诊断的患者以及年轻患者应进行该项检查。凡完全具备上述1~3项或1~4项者,可确诊。若高度怀疑该疾病,可考虑第5项检查。
2 结果
2.1 临床表现 症状:乏力9例(81.82%)、黄染9例(81.82%),纳差5例(45.45%),腹胀8例(72.73%),腹水8例(72.73%),水肿8例(72.73%),腹痛1例(9.09%),鼻衄1例(9.09%),步态不稳1例(9.09%),口齿不清1例(9.09%),头昏头痛1例(9.09%),抽搐1例(9.09%)。以神经系统症状为首发症状的2例(18.18%),以肝脏损害症状为首发症状的9例(81.82%)。体征:检查K-F环,阳性者9例(81.82%),其中3例有神经系统症状表现者K-F环均阳性,阳性率为100%。8例无神经系统表现者6例阳性,阳性率为75%。肝脏缩小的患者7例,脾大7例。巴氏征阳性1例。
2.2 实验室检查 血浆铜蓝蛋白:以<200 mg/L为诊断标准,<80 mg/L为诊断肝豆状核变性的有力证据[1]。11例患者均有血浆铜蓝蛋白不同程度的下降(20~140 mg/L),见表1。
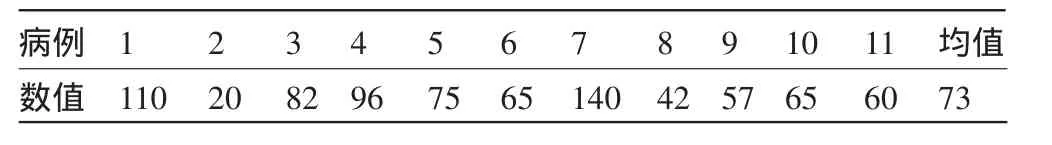
表1 11例患者血浆铜蓝蛋白值及均值(数值单位:mg/L)
2.3 影像学检查 腹部B超7例肝硬化、脾大,4例肝脏弥漫性损伤改变。10例头颅MRI中,6例无异常。2例双侧基底节区可见对称性分布信号。1例提示:双侧基底节区中脑变性病变。1例双侧基底节区、中脑异常信号影。其中3例有神经系统表现者均有异常信号。
3 讨 论
本病根据铜沉积部位的不同,临床表现多样,主要为肝脏和神经系统症状,肝脏病变多起病隐匿,表现为慢性病程,由乏力、厌食、黄疸、脾大,逐渐进展至门脉高压、肝性脑病、肝衰竭。神经系统病变以进行性的椎体外系运动障碍最为显著。本组病例肝损害达100%,高于既往文献报道[2],神经系统损害病例36.36%,低于既往文献报道[2],其原因可能与我科收治的患者多伴有肝脏损害有关。K-F环是肝豆状核变性相对特异性的体征,本组有神经系统症状患者均有K-F环,阳性率为100%,高于既往文献报道[3],考虑与病例偏少有关。本组病例血浆铜蓝蛋白均明显降低(100%),血浆铜蓝蛋白是肝脏合成的具有氧化酶活性的蛋白,85%~95%的肝豆状核变性患者血浆铜蓝蛋白浓度有不同程度的下降。因此,血浆铜蓝蛋白是肝豆状核变性的重要诊断指标[1]。肝功以及腹部影像学检查均可异常,但缺乏特异性。头颅MRI对中枢神经系统的改变程度很有意义,文献报道有60%神经系统症状患者和19%无神经系统症状患者,可通过MRI检测到脑部改变[4]。因其可能发现无神经系统症状患者的脑部病变,往往有助于诊断疾病。
肝豆状核变性患者是至今少数几种可治的神经遗传病之一,多数有症状的患者通过早期诊断,并终生药物治疗存活良好。故对该病早发现、早诊断尤为重要。为了能及时准确诊断肝豆状核变性疾病,需要对本病有充分认识。对原因不明的肝病和神经精神症状者均应警惕本病的可能,结合患者的临床症状及各种实验室、影像学检查结果正确分析避免误诊。
[1]丁晓东,杨 旭.肝豆状核变性的诊断和治疗[M]//范建高.非感染性肝病诊疗新进展2011-2012.北京:人民军医出版社,2011:59-62.
[2]姜 彬,王倩怡,谢琰臣,等.肝豆状核变性的诊断学进展[J].山东医药,2009,49(20):106-108.
[3]Ferenci P.pathophysiology and clinical features of Wilson disease[J].Metabolic Brain Disease,2004,19(3-4):229-239.
[4] Kitzberger R,Madl C,Ferenci P.Wilson Disease[J].Metabolic Brain Disease,2005,20(4):295-302.
