HPLC法测定复方芩兰口服液中连翘苷的含量
2012-07-28李延雪邵礼梅
李延雪 孙 菲 邵礼梅
黑龙江省鸡西市食品药品检验检测中心,黑龙江 鸡西 158100
复方芩兰口服液由金银花、黄芩、连翘、板蓝根四味中药经适宜加工方法制成的中药口服制剂,收载于《国家中成药标准汇编》内科肺系(一)分册[1],具有辛凉解表,清热解毒的功效,用于外感风热引起的发热、咳嗽、咽痛。据文献报道[2],连翘是一种广谱而有效的抗微生物药物,体外试验对许多种细菌有抑制作用,连翘对某些真菌亦有抑制作用。连翘苷作为连翘的主要成分,测定连翘苷的含量具有一定的意义。其原标准中没有连翘的含量测定方法和标准,为完善质量控制方法,参考相关文献[3-8]以连翘苷为测定指标,建立了复方芩兰口服液中连翘苷含量测定的高效液相色谱(HPLC)法,现报道如下:
1 仪器与试药
1.1 仪器
LC-2010A高效液相色谱仪(岛津):LC-2010紫外检测器,在线脱气机,四元泵,自动进样器,LC solution色谱工作站,色谱柱为 Agilent Extend-C18(4.6 mm×250 mm,5 μm);KQ-400KDE型高功效数控超声波清洗器,AG-285电子分析天平,TU-1901型双光束紫外可见光光度计(北京普析通用仪器有限责任公司)。
1.2 试药
对照品连翘苷(中国药品生物制品检定所,批号:110821-200609);复方芩兰口服液(批号:20110907、20110908、20110909);中性氧化铝(上海陆都化学试剂厂;粒度:100~200目;层析用);乙腈为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Extend-C18(4.6 mm×250 mm,5 μm);流动相:乙腈-水(25∶75);检测波长:278 nm;流速:1.0 mL/min;进样量:10 μL;柱温:30℃。理论板数按连翘苷峰计算不低于5000。
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取经减压干燥12 h的连翘苷对照品10.32 mg,置100 mL量瓶中,加50%甲醇约60 mL超声10 min,加50%甲醇至刻度,作为对照品溶液。
2.2.2 供试品溶液的制备 取本品5支混合均匀,精密量取本品 1 mL,加在中性氧化铝柱(粒度:100~200 目,6 g,内径为1 cm)上,用70%乙醇40 mL洗脱,收集洗脱液,将洗脱液置水浴上蒸干,残渣加50%甲醇3 mL,温热使溶解,转移至5mL量瓶中用50%甲醇稀释至刻度,摇匀,用微孔滤膜(0.45μm)滤过,取续滤液,即得供试品溶液。
2.2.3 阴性对照溶液的制备 取缺连翘的阴性样品,参照供试品溶液的制备方法,制得缺连翘的阴性对照溶液。
2.3 方法学考察
2.3.1 阴性干扰试验 分别精密吸取“2.2“项下3种溶液各10 μL,注入液相色谱仪。结果供试品溶液色谱图中,在与对照品溶液色谱相应位置上有色谱峰,阴性对照溶液色谱图中则无此峰,表明在该条件下,阴性样品不干扰连翘苷的测定,见图1。
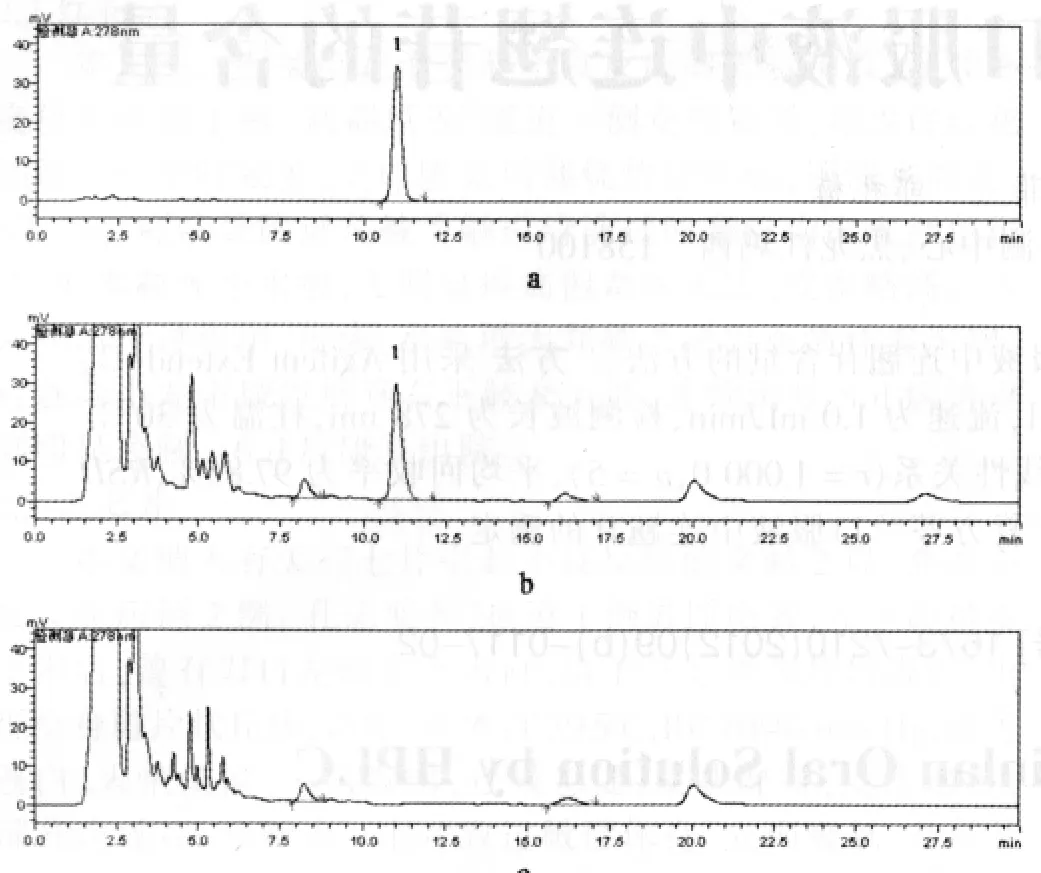
图1 复方芩兰口服液HPLC色谱图
2.3.2 线性关系考察 精密吸取上述对照品溶液2、5、10、15、20 μL,注入高效液相色谱仪,测定峰面积积分值,以峰面积积分值为纵坐标(Y),以对照品进样浓度为横坐标(X)绘制标准曲线, 得回归方程为 Y=6.2935×103X+1.6672×103,r=1.0000(n=5)。表明连翘苷在 20.64~206.40 μg/mL 范围内与峰面积值呈良好的线性关系。
2.3.3 精密度试验 精密吸取上述对照品溶液按上述色谱条件,连续进样5次,每次10 μL,测定连翘苷峰面积,连翘苷峰面积平均值为326389,RSD为0.74%(n=5),实验结果表明,在该条件下仪器精密度良好。
2.3.4 重现性试验 分别精密量取样品(批号:20110909)6份,按供试品溶液制备方法制备,依法测定,结果连翘苷的平均含量为 0.493 mg/mL,RSD=0.68%(n=6),表明方法重现性较好。
2.3.5 稳定性试验 精密吸取同一批供试品溶液 (批号:20110909),分别于配制后 0、2、4、6、8、12、24 h 进样,记录每次进样的峰面积。结果连翘苷的峰面积RSD=0.97%(n=7),表明供试品溶液在配制后24 h内基本稳定。
2.3.6 加样回收率试验 取已知含量的样品(批号:20110909,连翘苷含量为0.493 mg/mL)6份,每份精密量取本品0.5 mL,以两份为一组,共三组,每组分别精密加入连翘苷0.3955、0.4935、0.5920 mg,按供试品溶液的制备方法制备并测定含量,计算回收率,结果见表2。
2.4 样品含量测定
取市售三批样品,按供试品溶液的制备方法制备溶液,按拟定的色谱条件进样10 μL,测定峰面积,按外标法计算连翘苷的含量,结果见表3。
3 讨论
3.1 测定波长的选择
取连翘苷对照品适量,用50%甲醇稀释到一定的浓度,按照紫外分光光度法,在200~400 nm波长范围内扫描,连翘苷的最大吸收波长为278 nm,所以选择278 nm作为测定波长。
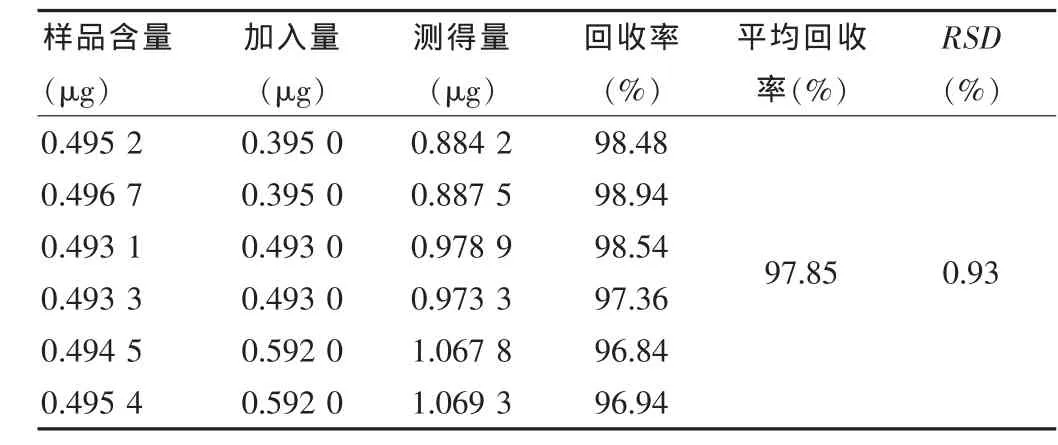
表2 加样回收率试验测定结果
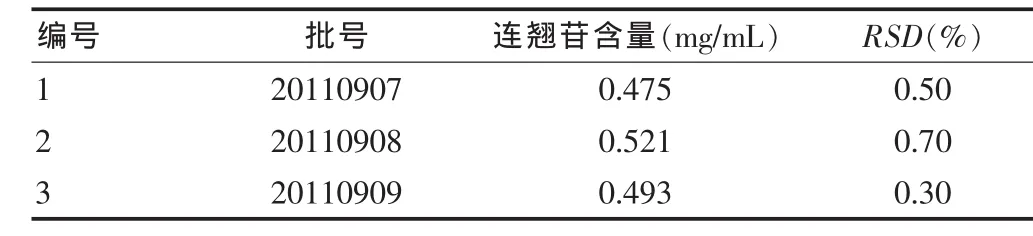
表3 样品含量测定结果(n=3)
3.2 供试品溶液的制备
曾采用50%的甲醇稀释,滤过,直接进样测定,结果连翘苷不能完全分离出来,干扰峰较多,参照《中国药典》2010年版一部[9],用中性氧化铝小柱处理样品,能有效去除杂质的干扰。
3.3 流动相的选择
为了达到良好的分离效果,曾采用不同浓度的甲醇-水、乙腈-水作为流动相进行分离,结果表明乙腈-水(25∶75)作为流动相,分离效果最好。
3.4 可用于含量测定
连翘作为复方芩兰口服液中的主要成分,测定连翘苷的含量具有一定的意义。本法灵敏度高,重复性好,可用于控制该制剂的质量。
[1]国家药品监督管理局.国家中成药标准汇编[S].内科肺系(一)分册,2002:217-219.
[2]南京中医药大学.中药大辞典(上册)[M].2版.上海:上海科学技术出版社,2006:1552-1555.
[3]张涛,陈学松.连翘不同炮制品中连翘苷的HPLC测定[J].中草药,2005,36(9):1339-1340.
[4]李亚荣.高效液相色谱法测定连翘败毒丸中连翘苷的含量[J].中国中医药杂志,2007,10(5):6-7
[5]韩桂茹,庞国勋.连翘苷检测波长的研究[J].中国药品标准,2005,6(3):71-73.
[6]索银科,祁小莉,罗兰,等.HPLC法测定退热解毒注射液中连翘苷的含量[J].中国药事,2010,24(10):1005-1007.
[7]陆红柳,谭喜莹,张飞,等.连翘中连翘苷HPLC测定方法的改进[J].中草药,2007,38(6):936-937.
[8]韩莉.连翘中连翘苷含量测定方法的试验研究[J].齐鲁药亊,2008,27(3):153-154.
[9]国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:611-612.
