紫草红素自乳化制剂的制备和初步质量评价
2012-07-26孙丹丹生立嵩徐新刚于蓓蓓闫雪生范秀丛
孙丹丹, 生立嵩, 徐新刚, 于蓓蓓, 闫雪生*, 范秀丛
(1.山东中医药大学,山东济南250355;2.山东省中医药研究院,山东济南250014)
自微乳化释药系统 (self-microemulsifying drug delivery system,SMEDDS)是由脂溶性或水难溶性药物、油相、乳化剂和助乳化剂组成,外观均一透明,在体外轻微振荡或体内胃肠道蠕动下,可自发乳化形成乳剂或微乳[1]。SMEDDS能有效地改善脂溶性药物在胃肠液中的溶解度和溶解速率[2],可以提高难溶性药物的口服生物利用度,促进药物的吸收[3]。SMEDDS在新剂型的应用方面拥有不可比拟的优势。
紫草为紫草科植物新疆紫草Arnebeia euchroma(Royle)Johnst.或内蒙紫草Arnebia guttata Bunge的干燥根,在《神农本草经》和《本草纲目》中均有记载,味甘、咸,寒,归心、肝经;功效清热凉血,活血解毒,透疹消斑[4]。近年来对于紫草红素及其衍生物的抗癌作用研究较多,可抑制多种肿瘤细胞的生长,促进肿瘤细胞凋亡,致肿瘤细胞周期阻滞[5]。连文员等[6]对左旋紫草素的自微乳制剂进行了研究,实验结果表明微乳对左旋紫草素有明显的增溶作用,为紫草素相关制剂的研究提供一定的依据。紫草红素水溶性差,临床应用时,由于其本身不易制剂,易被氧化,生物利用度低,因此本研究试将紫草红素研制成O/W型自微乳剂,以提高其溶解度。
1 材料
1.1 药品与试剂 紫草提取物 (自制);辛酸/癸酸三甘油酯 (GTCC)(浙江物美化学品有限公司);棕榈酸异丙酯 (IPP)、肉豆蔻酸异丙酯(IPM)(浙江物美化学品有限公司);油酸乙酯(上海飞祥化工厂);聚氧乙烯蓖麻油-35(EL-35)(德国BASF公司);Labrafil M 1944CS(法国GATTEFOSSE公司);聚乙二醇辛基苯基醚 (OP-10)(天津市广成化学试剂有限公司);Transcutol HP(法国GATTEFOSSE公司)。
1.2 仪器 Anke TDL—40B离心机 (上海安宁科学仪器厂);UV—265FW紫外-可见光分光光度计(岛津公司);AWM2000激光粒度分析仪、ZEN2600ZETA电位测定仪 (英国马尔文公司)。
2 方法与结果
2.1 紫草红素精制 取硅胶100 g湿法装柱,另取自制紫草提取物15 g,用乙酸乙酯溶解后硅胶拌样,挥干乙酸乙酯装柱,用石油醚-乙酸乙酯(20∶1)进行洗脱,合并洗脱液回收溶剂,得精制紫草红素,参照中国药典2010年版第320页紫草含量测定项下测定紫草红素含量为84.4%,转移率为90.7%。
2.2 紫草红素自微乳的制备
2.2.1 油水分配系数的测定 采用水—正辛醇法测定紫草红素在水和正辛醇中的浓度[7],分别测定其紫外吸光值,计算油水分配系数,制备方法及结果见表1。
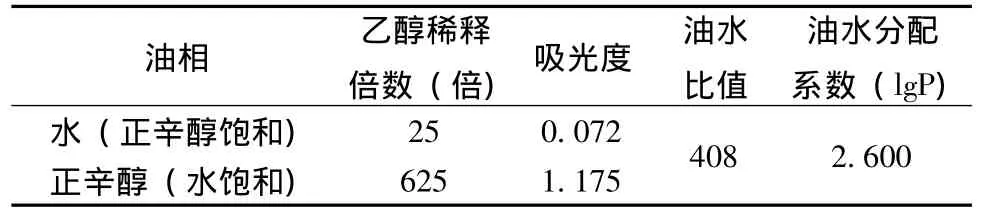
表1 油水分配系数测定结果Tab.1 Results of oil-water partition coefficient
由表1可知,紫草红素油水分配系数符合自微乳制剂制备的一般要求 (2<lgP<5),可将其制备成自微乳制剂。
2.2.2 紫草红素在不同油相中溶解度的考察 取精制紫草红素适量,精密称定质量,置具塞刻度试管中,加入一定量的油相 (GTCC、IPP、IPM、油酸乙酯),用90℃水浴使紫草红素完全分散于油相中成过饱和溶液,置室温下放凉,于4000 r/min,15 min离心,取上清液适量,用乙酸乙酯溶解定容,再用乙醇定量稀释一定倍数。按《中国药典》规定方法测定紫草红素含量并计算出溶解度,详见表2。
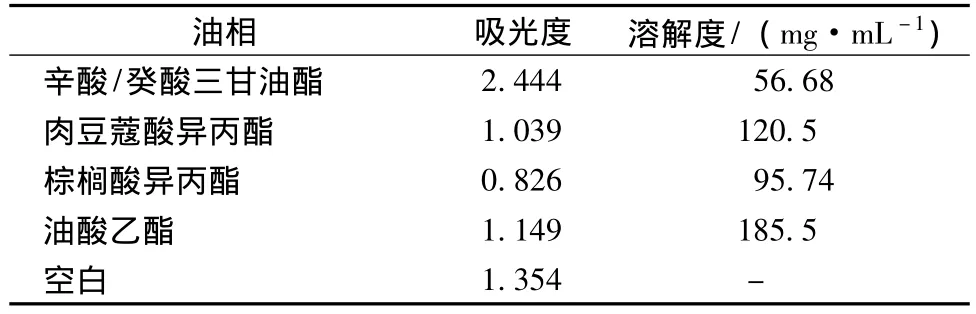
表2 紫草红素溶解度测定结果Tab.2 Results of red pigment shikonin's solubility in different oils
由上表结果可见,紫草红素于油酸乙酯中溶解度最大 (185.5 mg/mL),可以选择油酸乙酯做油相。
2.2.3 助乳化剂的筛选 在PEG—400、95%乙醇、Transcutol HP中筛选出一种助乳化剂,依次加入油酸乙酯、乳化剂 (吐温-80/聚氧乙烯蓖麻油-35=1∶1)、助乳化剂 (PEG—400、95%乙醇、Transcutol HP),固定油相用量,依次改变乳化剂和助乳化剂的用量,改变Km,后加入37℃水约80 mL将其稀释,观察现象,由结果可知,PEG—400所考察区域内无自乳化区,且分层较明显,Transcutol HP在所考察区域内虽然自乳化效果较好,形成透光性良好的蓝色乳光均一体系,但杯底有沉淀,说明此体系不够稳定,乙醇有透光性良好的区域,并澄清,不分层,选乙醇作为助乳化剂。
2.2.4 处方研究与筛选
2.2.4.1 不含药处方伪三元相图的绘制 分别取用不同组成和配比比例的乳化剂 (聚氧乙烯蓖麻油-35/聚乙二醇辛基苯基醚为1∶1、吐温-80/聚氧乙烯蓖麻油-35为1∶1、吐温-80/聚氧乙烯蓖麻油-35为3∶2、吐温-80/聚氧乙烯蓖麻油-35为2∶3、聚氧乙烯蓖麻油-35/Labrafil CS为2∶1)、油酸乙酯、95%乙醇,加入次序为油、乳化剂、助乳化剂,分别取Km=2、3、4、5、6、7、8中的若干个值,后用大量37℃恒温水稀释,观察记录稀释后的澄明度和乳光,绘制该系统的伪三元相图[8]。
由5个相图所描述出的自乳化区域可以看出自乳化区的区域大小稍有差别,因此,可以从不同乳化剂组合中筛选出自乳化区较大的乳化剂组合:聚氧乙烯蒽麻油-35/聚乙二醇辛基苯基醚=1∶1,吐温-80/聚氧乙烯蓖麻油-35=3∶2,分别从两相图中寻找乳化剂用量相对较少,而自乳化性状较好,即外观澄清或透明的系统 (图1)。
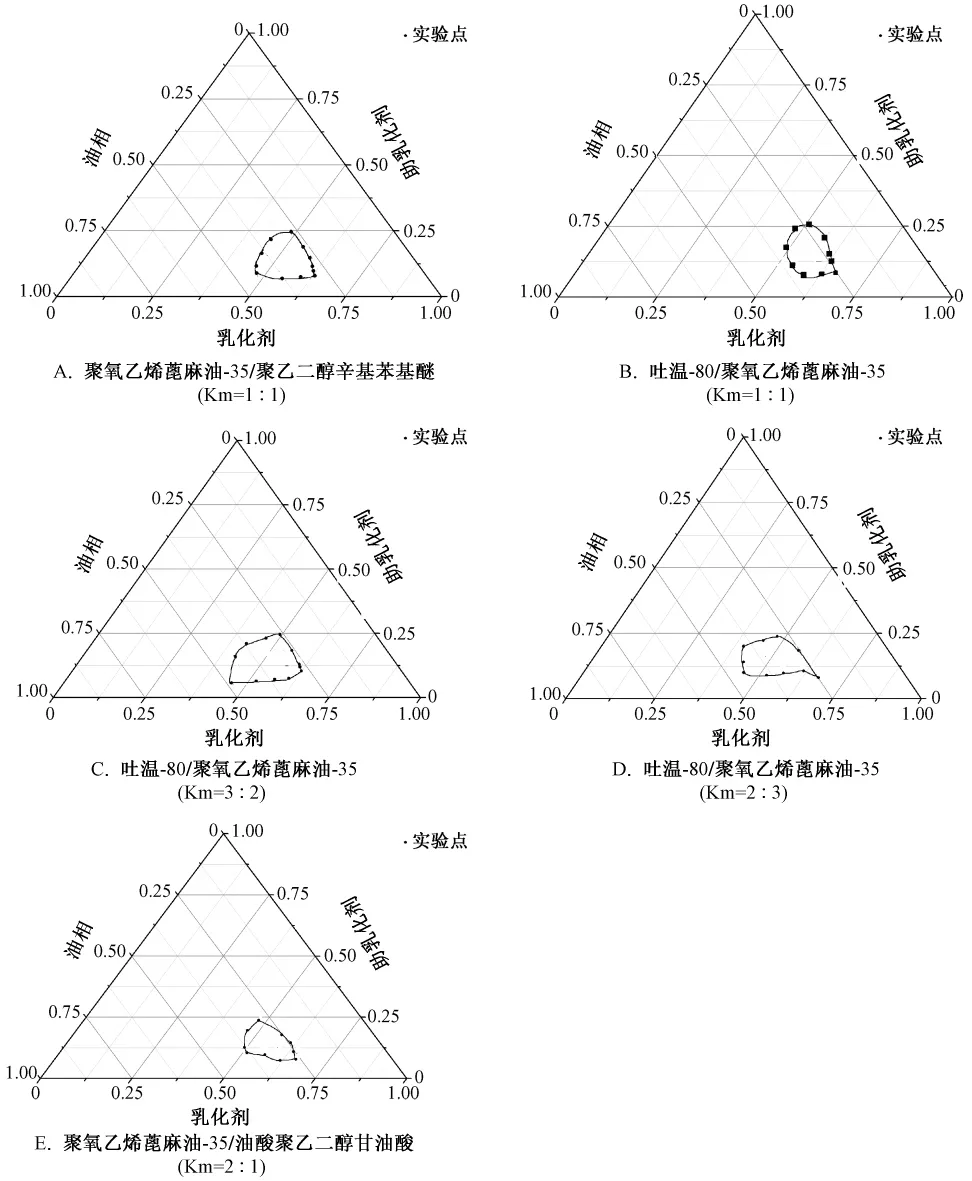
图1 不同组成和配比乳化剂的伪三元相图 (A~E)图中曲线所围区域为自微乳区Fig.1 Pseudo-ternary phase diagram of different emulsifiers(A -E).The region of SMEDDS is bound by the curved line
2.2.4.2 对不同乳化剂组合进行粒径测定 选择吐温-80/聚氧乙烯蓖麻油-35=3∶2(处方1~3)和聚氧乙烯蓖麻油-35/聚乙二醇辛基苯基醚=1∶1(处方4~6)为乳化剂、乙醇为助乳化剂、油酸乙酯为油相,制备如表3的空白自乳化制剂,采用马尔文公司的Master Sizer2000进行粒径测定,处方及结果见表3。
由表3可见,以吐温-80/聚氧乙烯蓖麻油-35=3∶2做乳化剂比聚氧乙烯蓖麻油-35/聚乙醇辛苯基醚=1∶1做乳化剂在空白自乳化中粒径小约40 nm左右,但空白处方的乳剂在与药物复合时会发生一定的相互作用,因此可以选出两者较优处方,进一步考察其载药处方粒径。

表3 空白自乳化制剂粒径结果Tab.3 Particle sizes of blank self-microemulsion preparations
2.2.4.3 含药处方粒径的测定及稳定性的初步考察 精密称取一定量药物和油酸乙酯,在50℃水浴加热的条件下使紫草红素完全溶解于油酸乙酯中,后依次加入定量的乳化剂和助乳化剂,使体系完全混溶,加37℃水使成自乳化体系,按上述方法测其粒径、Zeta电位,并通过长时间静置来观察其稳定性,处方及结果见表4。
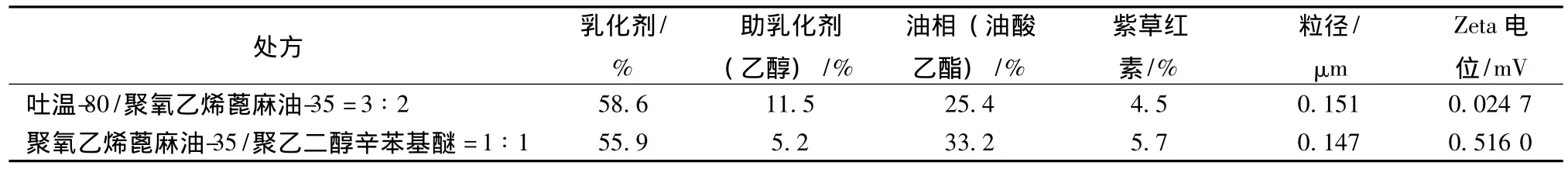
表4 Zeta电位测定结果Tab.4 Results of Zeta potential
Zeta电位由ZEN2600ZETA电位测定仪测得,直接测定的是电泳迁移率,并转化为Zeta电位,其两端为电极并且都施加了电势。粒子朝着相反电荷的电极运动,测量其速度并以单位场强表示,即其迁移率,进而转化为电位值,但由于药品有红蓝色的固有颜色,使得Zeta电位超出范围,因此只能通过长期静置来初步观察其稳定性,经观察系统均一稳定,未出现分层、沉淀现象,稳定性较好。陈小新等[9]对葛根素自微乳的研究也可说明微乳的稳定性并不是由 Zeta电位所决定的,而是由于表面活性剂的存在而导致的界面张力减小。
2.3 紫草红素自乳化固体颗粒的制备及质量的初步研究 以吐温-80/聚乙烯蓖麻油-35=3∶2为处方1的乳化剂,以聚乙烯蓖麻油-35/聚乙二醇辛基苯基醚=1∶1为处方2的乳化剂,用蔗糖粉和乳糖作为吸附剂,制颗粒。称取上述两种处方各1 g左右,渐加蔗糖、乳糖粉8 g、10 g、12 g做吸附剂,0.5 mL 50%乙醇做黏合剂制取颗粒,40℃烘干1 h,观察二者对于自乳化的吸附性能,并用Master sizer 2000测其粒径,处方及结果见表5,处方1和处方2用10倍量乳糖做吸附剂所得自微乳固体颗粒粒径测定结果如图2、图3。
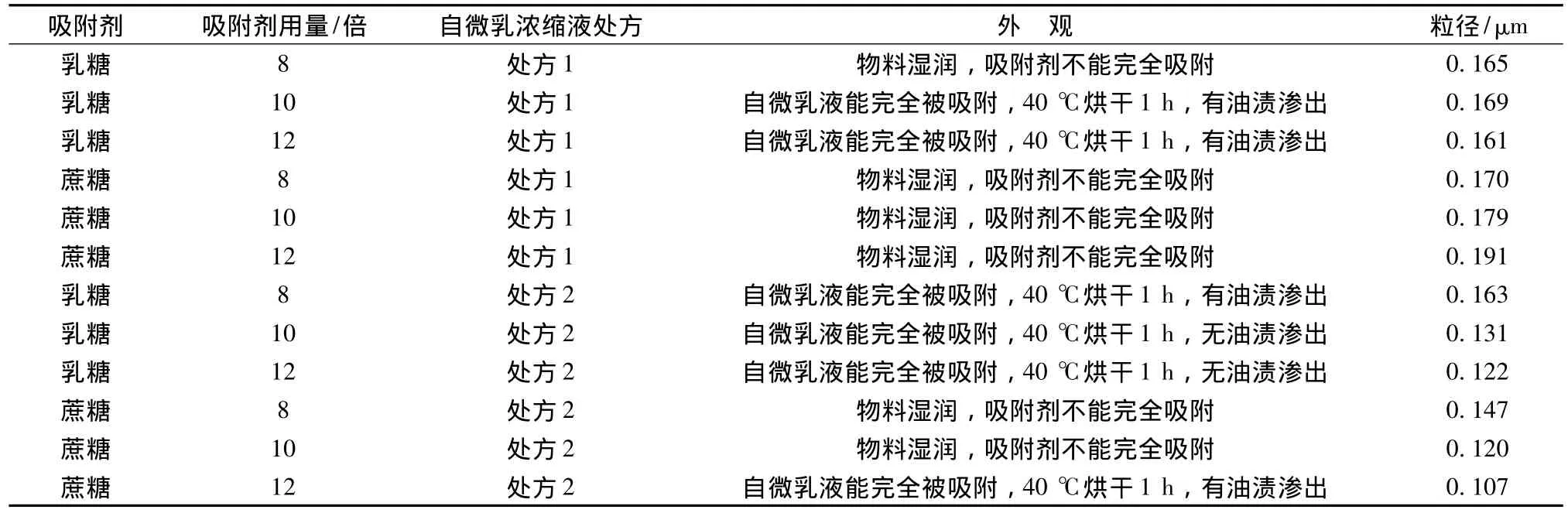
表5 含药自乳化制剂处方制备及粒径测定结果Tab.5 Preparation of drug-loaded SMEDDS and results of particle sizes
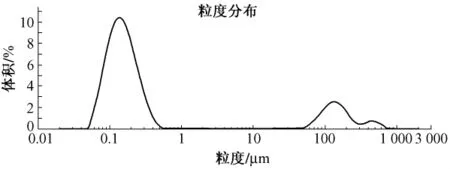
图2 处方1粒径测定图Fig.2 Particle size diagram of formula 1
可见,聚氧乙烯蓖麻油-35/聚乙二醇辛基苯基醚=1∶1做乳化剂时,使用10倍量乳糖吸附即可制得较理想的固体自乳化颗粒,且其自乳化速率及粒径较自乳化液体均无明显变化,因此可以选定此处方制备紫草红素固体自乳化制剂。
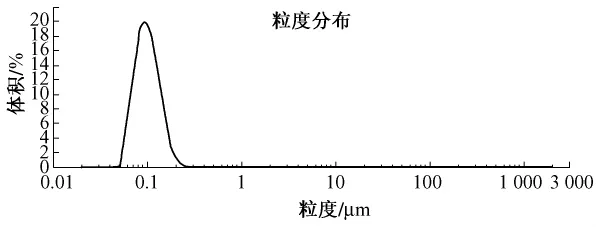
图3 处方2粒径测定图Fig.3 Particle size diagram of formula 2
3 讨论与结论
3.1 本实验对紫草红素自微乳处方进行了筛选,得到最优处方质量比为聚氧乙烯蓖麻油-35∶乳化剂聚乙二醇辛基苯基醚∶乙醇∶油酸乙酯∶紫草红素为28∶28∶5∶33∶6,以乳糖为固体吸附剂,用量为处方-乳糖 (1∶10 g/g)。
3.2 本实验得到的紫草红素自微乳粒径约为150 nm,粒径较大。但通过静置2个月来初步观察其稳定性,结果本释药系统均一稳定,未出现分层、沉淀现象,说明虽然粒径超过100 nm,但其稳定性较好。这可能是由于中药成分复杂,在微乳形成过程中不同成分会共同参与形成微乳结构,从而导致粒径偏大,但由于乳化剂的存在,影响微乳稳定性的界面张力并未增大,因此其稳定性也可达到预期目标。
3.3 本实验对蔗糖和乳糖作为紫草红素自乳化固体颗粒吸附剂进行了比较,乳糖吸附效果比较理想,对自微乳液的粒径影响不大。但固体吸附剂用量较大,其载药量受到限制,随着近年来新型脂质辅料和制药技术的发展,更多更好的固体自乳化技术不断涌现[10-12],今后可考虑对紫草红素自微乳的固体化技术进行进一步研究,以制备性质稳定,使用方便的固体自乳化制剂。
本实验对紫草素进行精制得到紫草红素,优选出了紫草红素自乳化系统的处方,粒径考察和稳定性考察结果表明本制剂粒径均一,符合自乳化要求,且稳定性良好,制备成固体颗粒载药量符合一般要求。
[1]Gursoy R N,Benita S.Self-emulsifying drug delivery systerms(SEDDS)for improved oral delivery of lipophilic drugs[J].Biomed Pharmacother,2004,58(3):173-182.
[2]凌 婧,孙明辉,翟雪珍,等.自 (微)乳化制剂的固体化技术[J].中国药学杂志,2011,46(4):245-248.
[3]袁海建,陈 彦,贾晓斌.中药自乳化药物传递系统的研究与应用[J].中华中医药杂志,2008,23(6):524-527.
[4]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:320.
[5]陈发奎.常用中草药有效成分含量测定[M].北京:人民卫生出版社,1973:732.
[6]连文元,李 悦,王新春,等.微乳对左旋紫草素增溶作用的实验研究[J].中成药,2010,32(5):879-881.
[7]齐雪萍,桂双英,鲁传华,等.汉防己甲素自微乳的研制[J].安徽医药,2010,14(4):399-401.
[8]潘国梁,贾晓斌,魏惠华,等.药用微乳伪三元相图的几种制备方法比较研究[J].中国药房,2006,17(1).
[9]陈小新,原 素,谢称石,等.葛根素自微乳给药系统的制备及其质量评价[J].中草药,2011,42(8):1512-1516.
[10]Balakrishnan P,Lee B T,Oh B H,et al.Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system(SEDDS)[J].Eur J Pharm,2009,72(3)539-545.
[11]Yi T,Wan J L,Xu H B,et al.A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water-soluble drugs[J].Eur J Pharm Biopharm,2008,70(2):439-444.
[12]Ito Y,Kusawake T,Ishida M,et al.Oral solid gentamicin preparation using emulsifier and adsorbent[J].J Controlled Release,2005,105(12):23-31.
